ORIGINAL PAPER Complete sequence and organisation of the Jatropha curcas (Euphorbiaceae) chloroplast genome Mehar H. Asif & Shrikant S. Mantri & Ayush Sharma & Anukool Srivastava & Ila Trivedi & Priya Gupta & Chandra S. Mohanty & Samir V. Sawant & Rakesh Tuli Received: 4 August 2009 / Revised: 17 April 2010 / Accepted: 6 May 2010 / Published online: 27 June 2010 # Springer-Verlag 2010 Abstract Jatropha curcas is an important non-edible oil seed tree species and is considered a promising source of biodiesel. The complete nucleotide sequence of J. curcas chloroplast genome (cpDNA) was determined by pyrosequencing and gaps filled by Sanger sequencing. The cpDNA is a circular molecule of 163,856 bp in length and codes for 110 distinct genes (78 protein coding, four rRNA and 28 distinct tRNA). Genome organisation and arrangement are similar to the reported angiosperm chloroplast genome. However, in Jatro- pha, the infA and the rps16 genes are non-functional. The inverted repeat (IR) boundary is within the rpl2 gene, and the 13 nucleotides at the ends of the two duplicate genes are different. Repeat analysis suggests the presence of 72 repeat regions (>30 bp) apart from the IR; of these, 48 were direct and 24 were palindromic repeats. Phylogenetic analysis of 81 protein coding chloroplast genes from 65 taxa by maximum parsimony, maximum likelihood and minimum evolution analyses at 100 bootstraps provide strong support for the placement of inaperturate crotonoids of which Jatropha is a member as sister to articulated crotonoids of which Manihot is a member. Keywords Jatropha curcas . Chloroplast . Genome . Phylogeny . Pyrosequencing . Euphorbiaceae . Angiosperms Introduction Jatropha curcas, a small tree or shrub belonging to the family Euphorbiaceae, is an important non-edible oil seed crop. It is a good source of oil since the seeds contains 25% to 40% fat by weight. The oil has a high calorific value and is therefore regarded as a potential fuel substitute (Jones and Miller 1991; Openshaw 2000). The plant shows wide adaptability, grows on marginal soils and can be multiplied rapidly both by seeds and cuttings. This has led to significant interest in improving it as a source of biodiesel (Jones and Miller 1991; Openshaw 2000). A number of studies are being undertaken to improve the various desirable traits of Jatropha like improvement of seed yield and oil quality and decreasing seed toxicity. Techniques to improve these traits include conventional breeding, creating interspecific hybrids, mutation breeding and genetic engineering (Sujatha et al. 2008). J. curcas is cultivated in tropical regions world- wide, especially in Asia and Africa, but has been suggested to be a native of America (King et al. 2009). Recent studies have shown that genetic diversity of J. curcas in Asia is very low (Basha and Sujatha 2007; Sudheer Pamidiamarri et al. 2009) which is consistent with cultivation far removed from its centre of origin. With so much interest in developing Jatropha for biofuel purposes, we undertook the sequencing of its chloroplast genome to study if it leads to new insights into the plant. Also since the lipid biosynthesis occurs in the chloroplast, it was an interesting option to see if any of the chloroplast genes show distinct variability. A total of 158 plastid genomes sequences have been reported in the National Centre for Biotechnology Infor- mation (NCBI) database out of which 132 are chloroplast genome sequences of green plants. Physical mapping and available sequence data revealed that chloroplast genomes Communicated by J. Dean Electronic supplementary material The online version of this article (doi:10.1007/s11295-010-0303-0) contains supplementary material, which is available to authorized users. M. H. Asif : S. S. Mantri : A. Sharma : A. Srivastava : I. Trivedi : P. Gupta : C. S. Mohanty : S. V. Sawant : R. Tuli (*) Plant Molecular Biology and Genetic Engineering Division, National Botanical Research Institute Council of Scientific and Industrial Research, Rana Pratap Marg, Lucknow 226001 Uttar Pradesh, India e-mail: [email protected]Tree Genetics & Genomes (2010) 6:941–952 DOI 10.1007/s11295-010-0303-0
Transcript
ORIGINAL PAPER
Complete sequence and organisation of the Jatropha curcas(Euphorbiaceae) chloroplast genome
Mehar H. Asif & Shrikant S. Mantri & Ayush Sharma &
Anukool Srivastava & Ila Trivedi & Priya Gupta &
Chandra S. Mohanty & Samir V. Sawant & Rakesh Tuli
Received: 4 August 2009 /Revised: 17 April 2010 /Accepted: 6 May 2010 /Published online: 27 June 2010# Springer-Verlag 2010
Abstract Jatropha curcas is an important non-edible oil seedtree species and is considered a promising source of biodiesel.The complete nucleotide sequence of J. curcas chloroplastgenome (cpDNA) was determined by pyrosequencing andgaps filled by Sanger sequencing. The cpDNA is a circularmolecule of 163,856 bp in length and codes for 110 distinctgenes (78 protein coding, four rRNA and 28 distinct tRNA).Genome organisation and arrangement are similar to thereported angiosperm chloroplast genome. However, in Jatro-pha, the infA and the rps16 genes are non-functional. Theinverted repeat (IR) boundary is within the rpl2 gene, and the13 nucleotides at the ends of the two duplicate genes aredifferent. Repeat analysis suggests the presence of 72 repeatregions (>30 bp) apart from the IR; of these, 48 were directand 24 were palindromic repeats. Phylogenetic analysis of 81protein coding chloroplast genes from 65 taxa by maximumparsimony, maximum likelihood and minimum evolutionanalyses at 100 bootstraps provide strong support for theplacement of inaperturate crotonoids of which Jatropha is amember as sister to articulated crotonoids of whichManihot isa member.
Jatropha curcas, a small tree or shrub belonging to thefamily Euphorbiaceae, is an important non-edible oilseed crop. It is a good source of oil since the seedscontains 25% to 40% fat by weight. The oil has a highcalorific value and is therefore regarded as a potentialfuel substitute (Jones and Miller 1991; Openshaw 2000).The plant shows wide adaptability, grows on marginalsoils and can be multiplied rapidly both by seeds andcuttings. This has led to significant interest in improving itas a source of biodiesel (Jones and Miller 1991; Openshaw2000). A number of studies are being undertaken toimprove the various desirable traits of Jatropha likeimprovement of seed yield and oil quality and decreasingseed toxicity. Techniques to improve these traits includeconventional breeding, creating interspecific hybrids,mutation breeding and genetic engineering (Sujatha et al.2008). J. curcas is cultivated in tropical regions world-wide, especially in Asia and Africa, but has beensuggested to be a native of America (King et al. 2009).Recent studies have shown that genetic diversity of J.curcas in Asia is very low (Basha and Sujatha 2007;Sudheer Pamidiamarri et al. 2009) which is consistentwith cultivation far removed from its centre of origin.
With so much interest in developing Jatropha for biofuelpurposes, we undertook the sequencing of its chloroplastgenome to study if it leads to new insights into the plant.Also since the lipid biosynthesis occurs in the chloroplast, itwas an interesting option to see if any of the chloroplastgenes show distinct variability.
A total of 158 plastid genomes sequences have beenreported in the National Centre for Biotechnology Infor-mation (NCBI) database out of which 132 are chloroplastgenome sequences of green plants. Physical mapping andavailable sequence data revealed that chloroplast genomes
Communicated by J. Dean
Electronic supplementary material The online version of this article(doi:10.1007/s11295-010-0303-0) contains supplementary material,which is available to authorized users.
M. H. Asif : S. S. Mantri :A. Sharma :A. Srivastava : I. Trivedi :P. Gupta : C. S. Mohanty : S. V. Sawant :R. Tuli (*)Plant Molecular Biology and Genetic Engineering Division,National Botanical Research Institute Council of Scientificand Industrial Research,Rana Pratap Marg,Lucknow 226001 Uttar Pradesh, Indiae-mail: [email protected]
Tree Genetics & Genomes (2010) 6:941–952DOI 10.1007/s11295-010-0303-0
of most land plants are highly conserved with respect totheir size, ranging from 120 to 217 kb (Ravi et al. 2006).The structure of the chloroplast genome is mainly con-served across land plants, with a pair of large invertedrepeat (IR) regions separated by the large single-copy(LSC) region and small single-copy (SSC) region. Theaverage number of genes encoded by the chloroplastgenome is 110–130 (Daniell et al. 2006). The genesencoded by the plastid genomes fall into three categories:(1) genetic system genes, including genes for rRNA, tRNA,ribosomal proteins and core subunits of eubacterial typeRNA polymerase; (2) photosynthesis-related genes; and (3)other conserved open reading frames (Shimada and Sugiura1991). The gene content and the polycistronic transcriptionunits of the chloroplast genomes of land plants are largelyconserved (Dixit et al. 1999; Kim and Lee 2004).
Monophyly of Euphorbiaceae has earlier been reportedbased on the molecular as well as embryological characters(Tokuoka 2007; Wurdack et al. 2005). The core Euphor-biaceae can be divided into seven major lineages (1)Erismantheae, (2) Acalyphoideae s.s., (3) Adenoclineaes.l., (4) Gelonieae, (5) articulated crotonoids, (6) inaperturatecrotonoids and (7) Euphorbioideae. Amongst the Euphorbia-ceae, the plastome sequence of Manihot has been reportedearlier (Daniell et al. 2008). Jatropha belongs to thesubfamily inaperturate crotonoids, whereas Manihot belongsto the articulated crotonoids (Tokuoka 2007).
The present study focuses on establishing the com-plete chloroplast genome sequence of J. curcas byanalysing and comparing it with the sequences availablein the database.
Materials and methods
Plant material
The J. curcas plant was obtained from the National Bureauof Plant Genetic Resources (Delhi, India). The accession ofthis species was NBPGR-RJ-Udi-0905-C-9. This plant wascollected from Udaipur region of Rajasthan (India).
DNA extraction and preparation of libraryfor pyrosequencing
Intact chloroplasts were prepared from J. curcas leaves bytwo-step percoll gradient method (Tanaka et al. 1987).DNA was extracted from the intact chloroplasts by phenol,chloroform and isoamyl alcohol method. The purified DNAwas precipitated by three volumes of absolute alcohol,pelleted, washed with 70% ethanol, air-dried and dissolvedin DNase-free water. This purified DNA was used forsequencing. For sequencing on 454 GSFLX sequencer, the
chloroplast DNA library was prepared, amplified andsequenced using the GS library preparation kit, GS emPCRkit1 and GS LR 70 sequencing kit, according to themanufacturer's protocol.
Sequencing, assembly and annotation
The J. curcas chloroplast genome was sequenced on 454GS FLX sequencer (Roche). DNA sequence data from theGS FLX sequencing run were assembled using version1.1.02.15 of the GS Assembler. A total of 102,003 readswith average length of 231.8 bases were assembled into 28contigs. The criteria for assembling the reads wereminimum overlap length of 40 bases with 90% identity.For ordering and orienting, the 28 contigs were mapped tochloroplast genome sequences of Arabidopsis and Populus.Some gaps were filled by fetching all the raw reads havingsequence similarity with contig ends. These reads werereassembled at 15 base overlap. The gaps which were filledby this method had homopolymer sequences of lengthgreater than seven bases in the gaps. The completedgenome sequence was annotated using DOGMA (Wymanet al. 2004). The genome map was constructed usingOGDraw version 1.1 (Lohse et al. 2007).
Primer design for filling gaps and RNA editing
After ordering and orienting of contigs, 28 gapsremained to be filled. Primers were designed from areasapproximately 100 bp away from each of the contigends, so that an amplification of approximately 200–300 bp was obtained for sequencing. The amplicons weresequenced by the Sanger's method, and the contig gapsfilled. For verification of the contigs assembly, primerswere designed from 11 regions of the chloroplast genomespanning a distance of 1 kb, and the amplicons weresequenced.
To validate the sites predicted for RNA editing in J.curcas, primers were designed from atpA, atpF, rps2,ndhB, clpP and accD genes. The primers used were:
Primer for rps2Forward: 5′GAGAATGGCACCTTATATCTCTGC 3′Reverse: 5′GTTTCTGTAGTGGACCAATTCGTTA 3′Primer for accDForward: 5′ACAAGCATTTGTGGGTTCAATG 3′Reverse: 5′CGATCTTTATAAGGTTCCTCTTCTG 3′Primer for atpFForward: 5′GTAACCGATTCTTTCGTTTCCTT 3′Reverse: 5′ATCTTTCCATTCATTGCAAAACTT 3′Primers for ndhBForward: 5′CGATGGAGAGAAGAACCTATGATT 3′Reverse: 5′TTGATATTCCCGGTATAGTAGATGC 3′
942 Tree Genetics & Genomes (2010) 6:941–952
Primers for clpP1Forward: 5′ATTCCCTCATGCTAGGGTAATGAT 3′Reverse: 5′CAACTGCTACAAGGTCAACAATTC 3′Primer for atpAForward: 5′CGCGTAATCGTTGACCTCTT 3′Reverse: 5′GTACCGGGAACGACACACTT 3′
For the reverse transcription reaction, total RNA wasisolated from Jatropha leaves and treated with RNase-freeDNase. The reverse transcription (RT) reaction was carriedout by the Invitrogen SuperscriptII reverse transcriptaseaccording to the manufacturer's protocol. For the RTreaction, the reverse primers for each gene were used tomake the single-stranded cDNA. The PCR was carried outfrom an aliquot of the RT reaction using the Invitrogen TaqDNA polymerase and primers specific for each genes. Theamplicons obtained were sequenced using the ABI DNAanalyser 3730xl.
Repeat analysis
To identify the repeat regions in the Jatropha chloroplastgenome, the programme REPuter (Kurtz et al. 2001) wasused to identify the number and location of direct andinverted repeats using a minimum repeat size of 30 bp andhamming distance of 3.
Phylogeny and whole-genome alignments
For the phylogenetic analysis, complete data matrix whichincludes 81 genes from 64 taxa (Jansen et al. 2007) wasused in which the Jatropha sequences were added. Allpositions containing gaps and missing data were eliminatedfrom the dataset (complete deletion option). The sequenceswere aligned using ClustalW (Higgins et al. 1996), andmaximum parsimony (MP) tree and minimum evolution(ME) tree were made in MEGA 4 software (Tamura et al.2007). For ML analysis, the GARLI0.96 software was used(Zwickl 2006) at the automated stopping criterion, termi-nating the search when the –ln score remained constant for20,000 consecutive generations. For each analysis, 100bootstrap (BS) replicates were used.
For ME analysis, the evolutionary distances werecomputed using the Nei–Gojobori method (Nei andGojobori 1986) and are in the units of the number ofsynonymous differences per sequence. The ME tree wassearched using the close-neighbour interchange (CNI)algorithm (Nei and Kumar 2000) at a search level of 3.
The MP tree was obtained using the close-neighbourinterchange algorithm (Nei and Gojobori 1986) with searchlevel 3 (Nei and Gojobori 1986) in which the initial treeswere obtained with the random addition of sequences (51replicates).
The tree was viewed manipulated using TreeView(Page 1996).
Results
Sequence assembly and annotation
The 102,003 reads that were obtained on sequencing wereassembled into 28 contigs of an average length of 4.8 kband the largest contig length of 20.3 kb (Table 1). Afterassembly, the contigs were mapped to the chloroplastgenome of Arabidopsis and Populus for ordering andorienting. After ordering and orienting, 28 gaps remainedto be filled. Primers were designed for all the 28 gaps asdescribed in “Materials and methods”. However, nine gapswere also filled by fetching the raw reads that had similarityto the ends of the contigs and then assembled at criteria of15-bp overlap with over 90% identity. Except for the IRa(rpl2)-trnH gap which was >300 bp, all the other gaps wereless than 100 bp, most of the gaps that were filled bysequencing were homopolymer regions. To check for thecorrect assembly of the reads, primers were designed for 11regions including the protein coding genes and intergenicregions. These amplicons were sequenced and aligned tothe assembled sequence. In all the cases, there was no casein which any discrepancy in assembly or sequencing wasdetected. The overall coverage of the chloroplast genome(including both the IR) was 145×. The complete sequencethus obtained was annotated with DOGMA (Wyman et al.2004).
Genome arrangement and structure
The complete nucleotide sequence of the chloroplastgenome of J. curcas was determined to be 163,856 bp(GenBank accession no. FJ695500; RefSeq numberNC_012224). It includes a pair of inverted repeats (IRaand IRb) of 27,124 bp, large single-copy (LSC) region of
Table 1 Characteristics of the GS FLX run data assembly
Size of largest contig 20,368
Total no. of reads 102,003
Average read length 231.8
Overall average read depth (incl. one IR) 174.4567
Overall average read depth (incl. both IRs) 145.3209
Inverted Repeat average read depth 281.2173
Single-copy region average read depth 147.6843
Proportion of bases≥Q40 86.76%
No. of gaps 28
Tree Genetics & Genomes (2010) 6:941–952 943
91,756 bp and SSC region of 17,852 bp, respectively(Fig. 1). The Jatropha chloroplast genome encodes 130genes, of which 91 are single copy and 17 are duplicated inthe IR region (Table 2). There are four distinct rRNA genes,28 distinct tRNA and 78 distinct protein coding genes. Ofthese, six protein coding, four rRNA and seven tRNA genesare duplicated in the IR, and two tRNA genes (trnG-GCCand trnM-CAU) are present in two copies in the single-copyregion. A part of the ycf1 gene is also present as apseudogene. The AT content of the genome is 64.64% andGC content is 35.36%. The genome consists of 50.2%protein coding, 5.5% rRNA coding, 1.7% tRNA and 42.6%non-coding sequences. There are 15 intron-containinggenes, of which three (clpP, ycf3 and rps12) have twointrons and the rest have one intron each. Four intron-containing genes (ndhA, rps12_3 end, rpl2 and trnA) areduplicated in the IR.
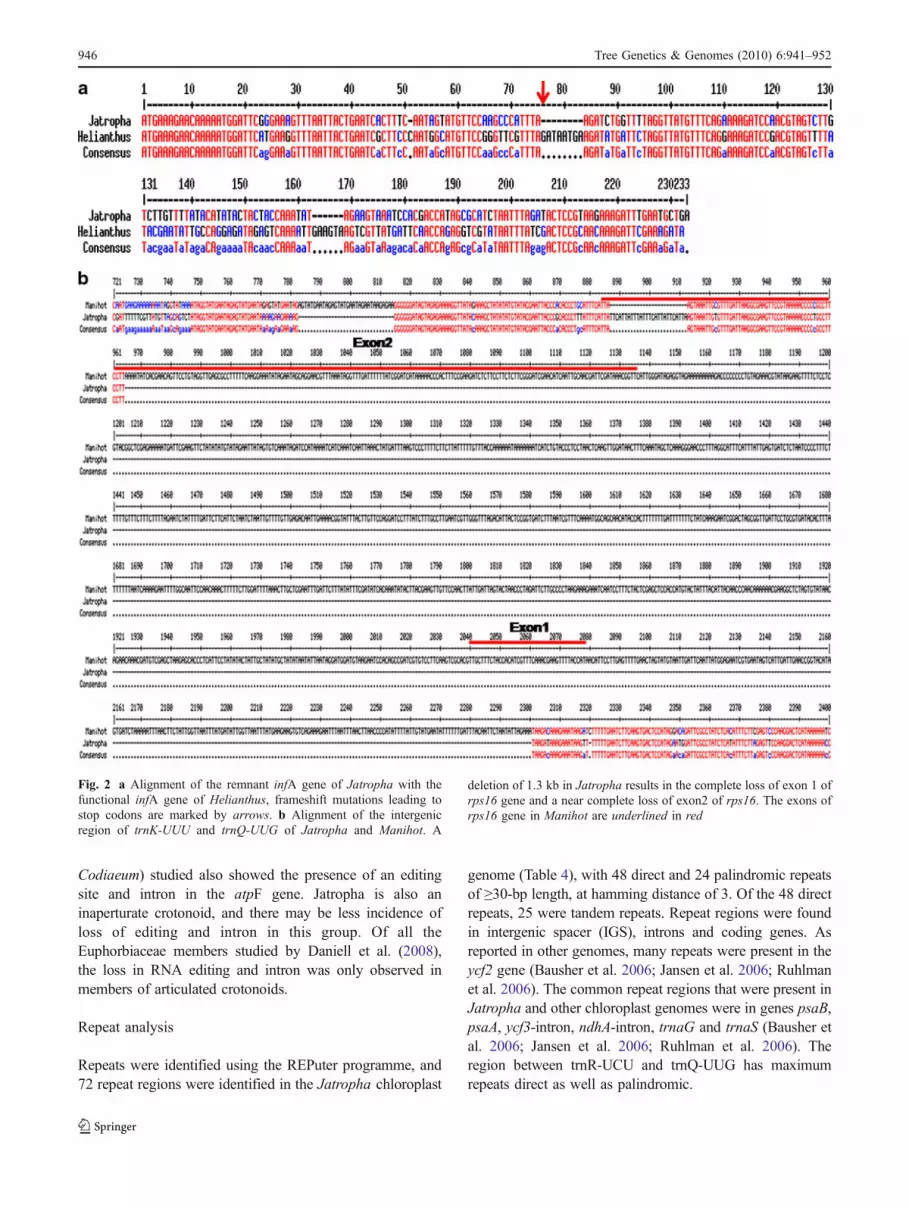
The infA gene coding for the translation initiation factor1 and rps16 gene coding for the small subunit ribosomalprotein are non-functional in the Jatropha chloroplastgenome. Similar to the other rosids, a remnant of the infAgene is present in the intergenic region of rpl36 and rps8and is characterised by multiple frameshift mutations(Fig. 2a).
The rps16 gene is nearly completely missing because ofa 1.3-kb deletion in the intergenic region of the trnK-UUUand trnQ-UUG which results in the complete loss of exon 1of rps16 and near complete loss of exon 2 of rps16(Fig. 2b). This deletion has also been observed in 14 of the32 chloroplast genomes lacking rps16.
The boundary of the IR and LSC resides within the rpl2gene; the last 13 nucleotides of the rpl2 gene in IRa andIRb are different; the IR and SSC boundary is at the ycf1gene. However, the partial ycf1 gene in the IRb is larger
Fig. 1 Gene map of the J. curcas chloroplast genome. The thick linesindicate the inverted repeats (IRa and IRb), which separate thegenome into small (SSC) and large (LSC) single-copy regions. Genes
on the outside of the map are transcribed in the clockwise directionand genes on the inside of the map are transcribed in counter-clockwise direction
944 Tree Genetics & Genomes (2010) 6:941–952
than that reported from other genomes (2.2 kb as comparedto generally 1 kb). Apart from the annotated genes, we alsoidentified an open reading frame (ORF) in the IR regionORF126. It codes for a 126 aa protein, and this region ishighly conserved in 73 of the 133 chloroplast genome IRregions.
Insertions specific to Jatropha chloroplast genome arepresent in the matK and ndhF genes. In the matK gene, a 9-bp insertion at position 204 bp is present in Jatropha and a6-bp insertion at position 1,071 bp is present in bothJatropha and Manihot. This 6-bp insertion in matK may bespecific to Euphorbiaceae as it is not present in any otherchloroplast genomes.
In the ndhF gene, a 12-bp insertion at position 1,930 bpis present in Jatropha. All the Jatropha-specific insertionsare mainly AT rich; these insertions account for the largersize of the Jatropha chloroplast genome and a slightlyhigher AT content when compared to Manihot.
Atypical start codons, ACG and GTG, were observed inndhD and rps19, respectively. RNA editing has been welldocumented for the ndhD gene in other plant species(Tsudzuki et al. 2001). A C/T editing occurs in ndhD startcodon, changing it from ACG to ATG. Forty-three RNA
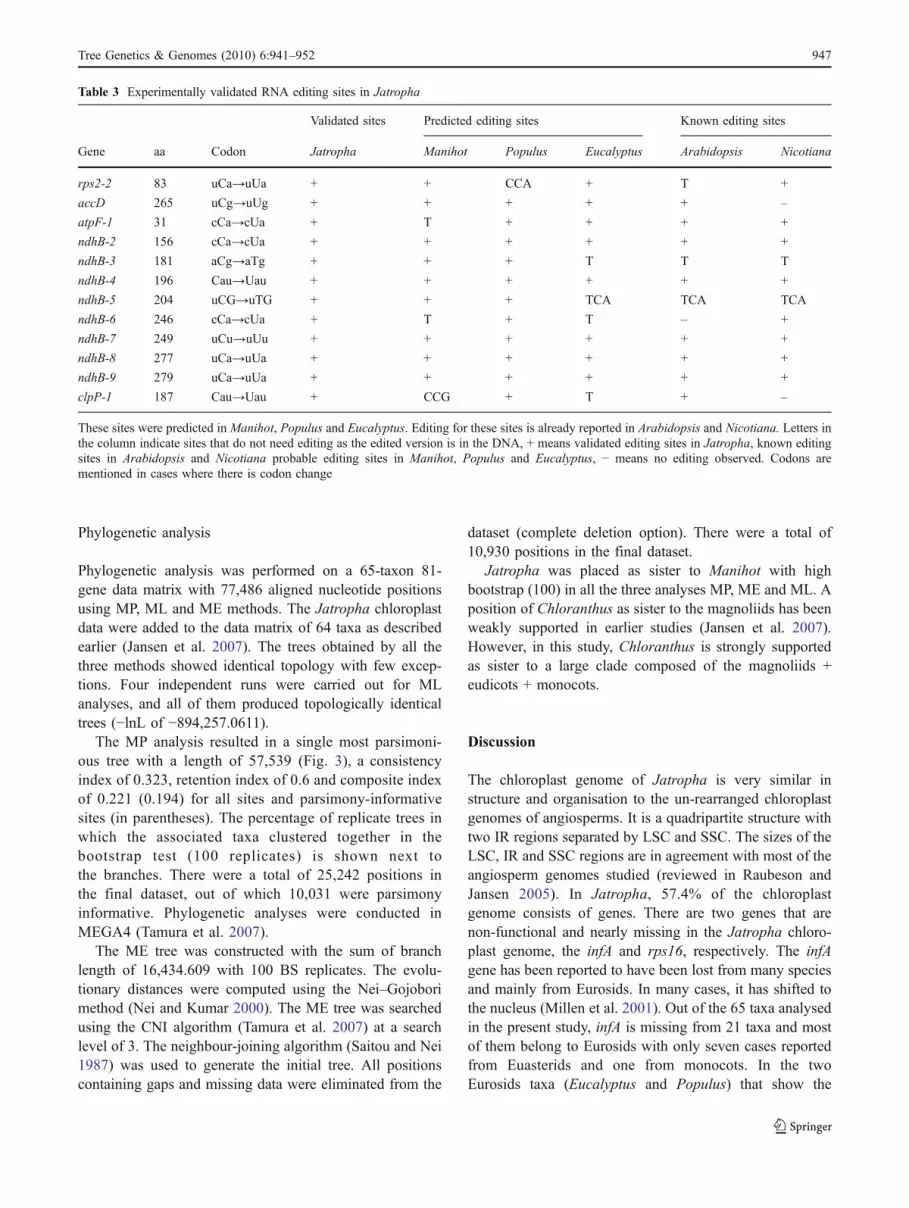
editing sites studied in Arabidopsis and Nicotiana werechecked for the probable editing in Jatropha, Manihot,Populus and Eucalyptus (Supplementary Table 1). Out of43 sites, Jatropha, Manihot, Populus and Eucalyptus hadedited DNA versions at 8, 12, 9 and 16 sites, codon changeat 5, 2, 4 and 0 sites and probable editing at 28, 26, 26 and24 sites, respectively. The probable editing sites of atpA,atpF, rps2, ndhB, clpP and accD were checked bysequencing the RTPCR products. All the editing sitespredicted earlier in these genes were validated bysequencing (Table 3). A total of 12 editing sites werevalidated, and all of them were non-synonymous substitu-tions. There was no case where a predicted editing site wassequenced and no editing found.
As observed by Daniell et al. (2008), the editing in atpFgene at nucleotide position 92, from C to T, and the presenceof an intron associated in Malpighiales was also substanti-ated in the present study. The editing in atpF gene atnucleotide position 92 from C to T was also observed inJatropha, along with the presence of an intron. Interestingly,many cases of RNA editing in the intron of the unprocessedtranscript of atpF was also observed. In the study by Daniellet al. (2008), the two inaperturate crotonoids (Croton,
Table 2 Genes present in the Jatropha chloroplast genome
Subunits of Photosystem I psaA, psaB, psaC, psaI, psaJ
Subunits of Photosystem II psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ
Large subunit of Rubisco rbcL
Subunits of cytochrome b/fcomplex
petA, petB, petD, petG, petL, petN
Subunits of ATP synthase atpA, atpB, atpE, atpFb, atpH, atpI
Cytochrome c biogenesis ccsA
Maturase matK
Acetyl –CoA carboxylase accD
Protease clpPb
Envelope membrane protein cemA
Conserved hypotheticalgenes
ycf1a, ycf2a, ycf3b, ycf4,orf126
a Genes present in the IR regionsb Genes having introns
Tree Genetics & Genomes (2010) 6:941–952 945
Codiaeum) studied also showed the presence of an editingsite and intron in the atpF gene. Jatropha is also aninaperturate crotonoid, and there may be less incidence ofloss of editing and intron in this group. Of all theEuphorbiaceae members studied by Daniell et al. (2008),the loss in RNA editing and intron was only observed inmembers of articulated crotonoids.
Repeat analysis
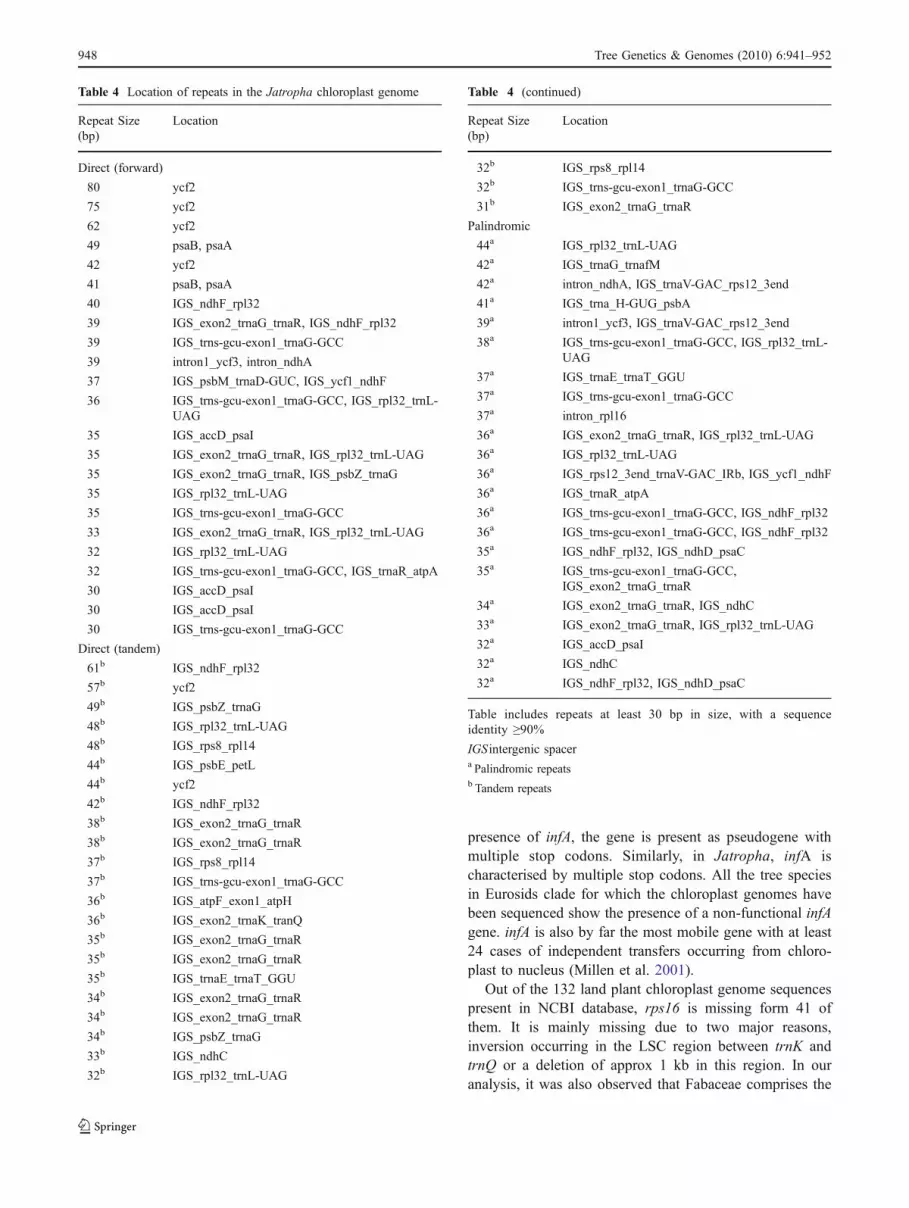
Repeats were identified using the REPuter programme, and72 repeat regions were identified in the Jatropha chloroplast
genome (Table 4), with 48 direct and 24 palindromic repeatsof ≥30-bp length, at hamming distance of 3. Of the 48 directrepeats, 25 were tandem repeats. Repeat regions were foundin intergenic spacer (IGS), introns and coding genes. Asreported in other genomes, many repeats were present in theycf2 gene (Bausher et al. 2006; Jansen et al. 2006; Ruhlmanet al. 2006). The common repeat regions that were present inJatropha and other chloroplast genomes were in genes psaB,psaA, ycf3-intron, ndhA-intron, trnaG and trnaS (Bausher etal. 2006; Jansen et al. 2006; Ruhlman et al. 2006). Theregion between trnR-UCU and trnQ-UUG has maximumrepeats direct as well as palindromic.
Fig. 2 a Alignment of the remnant infA gene of Jatropha with thefunctional infA gene of Helianthus, frameshift mutations leading tostop codons are marked by arrows. b Alignment of the intergenicregion of trnK-UUU and trnQ-UUG of Jatropha and Manihot. A
deletion of 1.3 kb in Jatropha results in the complete loss of exon 1 ofrps16 gene and a near complete loss of exon2 of rps16. The exons ofrps16 gene in Manihot are underlined in red
946 Tree Genetics & Genomes (2010) 6:941–952
Phylogenetic analysis
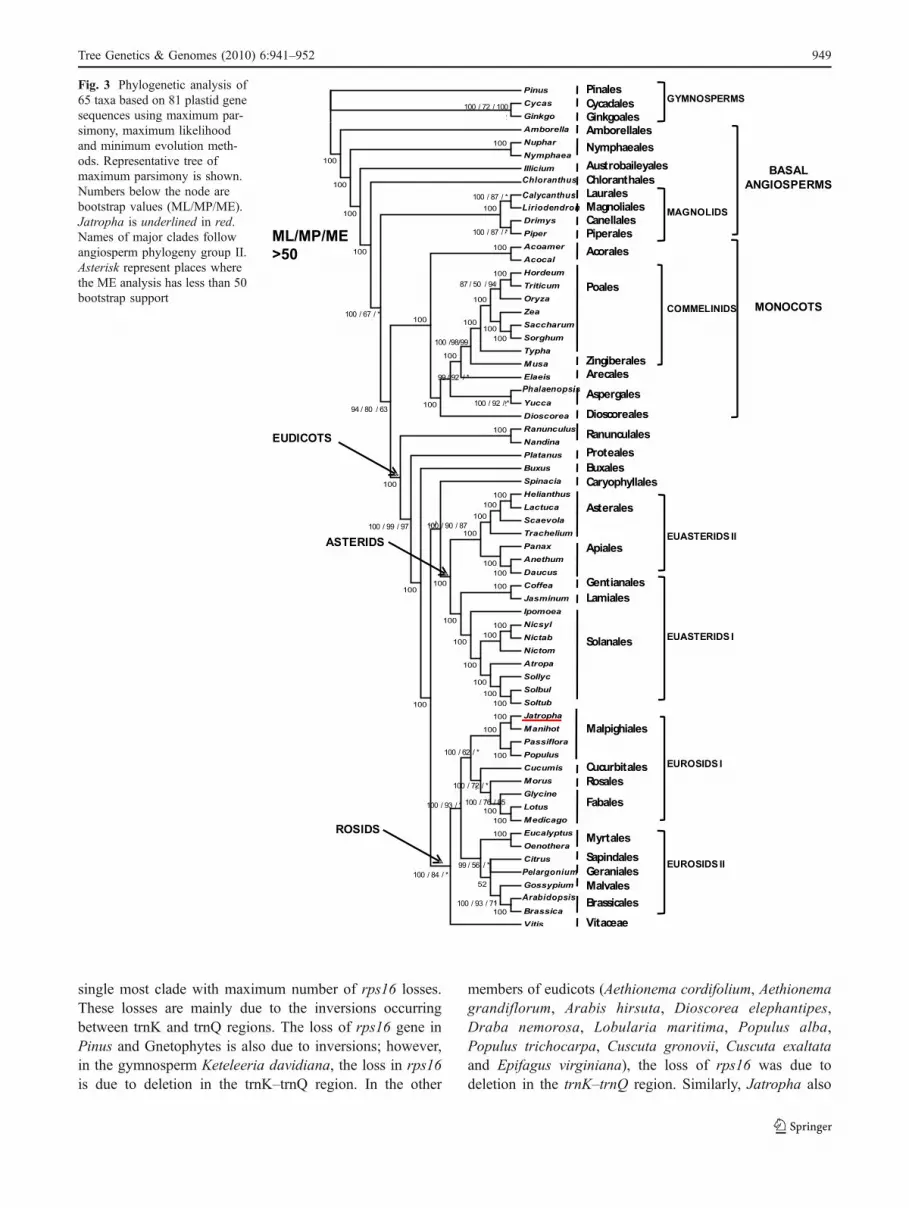
Phylogenetic analysis was performed on a 65-taxon 81-gene data matrix with 77,486 aligned nucleotide positionsusing MP, ML and ME methods. The Jatropha chloroplastdata were added to the data matrix of 64 taxa as describedearlier (Jansen et al. 2007). The trees obtained by all thethree methods showed identical topology with few excep-tions. Four independent runs were carried out for MLanalyses, and all of them produced topologically identicaltrees (−lnL of −894,257.0611).
The MP analysis resulted in a single most parsimoni-ous tree with a length of 57,539 (Fig. 3), a consistencyindex of 0.323, retention index of 0.6 and composite indexof 0.221 (0.194) for all sites and parsimony-informativesites (in parentheses). The percentage of replicate trees inwhich the associated taxa clustered together in thebootstrap test (100 replicates) is shown next tothe branches. There were a total of 25,242 positions inthe final dataset, out of which 10,031 were parsimonyinformative. Phylogenetic analyses were conducted inMEGA4 (Tamura et al. 2007).
The ME tree was constructed with the sum of branchlength of 16,434.609 with 100 BS replicates. The evolu-tionary distances were computed using the Nei–Gojoborimethod (Nei and Kumar 2000). The ME tree was searchedusing the CNI algorithm (Tamura et al. 2007) at a searchlevel of 3. The neighbour-joining algorithm (Saitou and Nei1987) was used to generate the initial tree. All positionscontaining gaps and missing data were eliminated from the
dataset (complete deletion option). There were a total of10,930 positions in the final dataset.
Jatropha was placed as sister to Manihot with highbootstrap (100) in all the three analyses MP, ME and ML. Aposition of Chloranthus as sister to the magnoliids has beenweakly supported in earlier studies (Jansen et al. 2007).However, in this study, Chloranthus is strongly supportedas sister to a large clade composed of the magnoliids +eudicots + monocots.
Discussion
The chloroplast genome of Jatropha is very similar instructure and organisation to the un-rearranged chloroplastgenomes of angiosperms. It is a quadripartite structure withtwo IR regions separated by LSC and SSC. The sizes of theLSC, IR and SSC regions are in agreement with most of theangiosperm genomes studied (reviewed in Raubeson andJansen 2005). In Jatropha, 57.4% of the chloroplastgenome consists of genes. There are two genes that arenon-functional and nearly missing in the Jatropha chloro-plast genome, the infA and rps16, respectively. The infAgene has been reported to have been lost from many speciesand mainly from Eurosids. In many cases, it has shifted tothe nucleus (Millen et al. 2001). Out of the 65 taxa analysedin the present study, infA is missing from 21 taxa and mostof them belong to Eurosids with only seven cases reportedfrom Euasterids and one from monocots. In the twoEurosids taxa (Eucalyptus and Populus) that show the
Table 3 Experimentally validated RNA editing sites in Jatropha
Validated sites Predicted editing sites Known editing sites
Gene aa Codon Jatropha Manihot Populus Eucalyptus Arabidopsis Nicotiana
rps2-2 83 uCa→uUa + + CCA + T +
accD 265 uCg→uUg + + + + + –
atpF-1 31 cCa→cUa + T + + + +
ndhB-2 156 cCa→cUa + + + + + +
ndhB-3 181 aCg→aTg + + + T T T
ndhB-4 196 Cau→Uau + + + + + +
ndhB-5 204 uCG→uTG + + + TCA TCA TCA
ndhB-6 246 cCa→cUa + T + T – +
ndhB-7 249 uCu→uUu + + + + + +
ndhB-8 277 uCa→uUa + + + + + +
ndhB-9 279 uCa→uUa + + + + + +
clpP-1 187 Cau→Uau + CCG + T + –
These sites were predicted in Manihot, Populus and Eucalyptus. Editing for these sites is already reported in Arabidopsis and Nicotiana. Letters inthe column indicate sites that do not need editing as the edited version is in the DNA, + means validated editing sites in Jatropha, known editingsites in Arabidopsis and Nicotiana probable editing sites in Manihot, Populus and Eucalyptus, − means no editing observed. Codons arementioned in cases where there is codon change
Tree Genetics & Genomes (2010) 6:941–952 947
presence of infA, the gene is present as pseudogene withmultiple stop codons. Similarly, in Jatropha, infA ischaracterised by multiple stop codons. All the tree speciesin Eurosids clade for which the chloroplast genomes havebeen sequenced show the presence of a non-functional infAgene. infA is also by far the most mobile gene with at least24 cases of independent transfers occurring from chloro-plast to nucleus (Millen et al. 2001).
Out of the 132 land plant chloroplast genome sequencespresent in NCBI database, rps16 is missing form 41 ofthem. It is mainly missing due to two major reasons,inversion occurring in the LSC region between trnK andtrnQ or a deletion of approx 1 kb in this region. In ouranalysis, it was also observed that Fabaceae comprises the
Table 4 Location of repeats in the Jatropha chloroplast genome
Table includes repeats at least 30 bp in size, with a sequenceidentity ≥90%IGS intergenic spacera Palindromic repeatsb Tandem repeats
948 Tree Genetics & Genomes (2010) 6:941–952
single most clade with maximum number of rps16 losses.These losses are mainly due to the inversions occurringbetween trnK and trnQ regions. The loss of rps16 gene inPinus and Gnetophytes is also due to inversions; however,in the gymnosperm Keteleeria davidiana, the loss in rps16is due to deletion in the trnK–trnQ region. In the other
members of eudicots (Aethionema cordifolium, Aethionemagrandiflorum, Arabis hirsuta, Dioscorea elephantipes,Draba nemorosa, Lobularia maritima, Populus alba,Populus trichocarpa, Cuscuta gronovii, Cuscuta exaltataand Epifagus virginiana), the loss of rps16 was due todeletion in the trnK–trnQ region. Similarly, Jatropha also
Fig. 3 Phylogenetic analysis of65 taxa based on 81 plastid genesequences using maximum par-simony, maximum likelihoodand minimum evolution meth-ods. Representative tree ofmaximum parsimony is shown.Numbers below the node arebootstrap values (ML/MP/ME).Jatropha is underlined in red.Names of major clades followangiosperm phylogeny group II.Asterisk represent places wherethe ME analysis has less than 50bootstrap support
Tree Genetics & Genomes (2010) 6:941–952 949
had a 1.3-kb deletion in the trnK–trnQ region, resulting inthe loss of the rps16 gene. Its closest relative Manihot had afunctional rps16 gene. No loss of rps16 from monocots hasbeen reported
It has been shown for Populus and Medicago that loss ofchloroplast rps16 is compensated by a mitochondrial rps16which has dual targeting activity and also functions in thechloroplast (Ueda et al. 2008). This could also hold true forother species where the rps16 gene is missing.
Apart from the missing genes, an ORF126 was presentin the Jatropha chloroplast genome in the IR region. TheORF126 encodes a hypothetical protein of 126 aa, and thesequence is conserved in 73 of the 133 chloroplast genomesanalysed by MUMMER (Kurtz et al. 2004), and thesimilarity ranges from 75% to 95%.
Repeat analyses identified many direct and palindromicrepeat regions. A large number of the repeats were presentin the IGS. The repeats that were present in the codingregions were similar to those reported in the other genomes(Bausher et al. 2006; Jansen et al. 2006; Ruhlman et al.2006). Apart from the IR, the largest repeat region was of80 bp which is a part of the ycf2 gene. The large number ofrepeats could be a result of the expanding chloroplastgenome. In Jatropha, the IRb-SSC boundary has expandedto include a larger part of the ycf1 gene, generally, thepartial ycf1 in the IR is found to be in the range of 156 to1,583 bp (Raubeson et al. 2007). However, in Jatropha, thepartial ycf1 gene is of 2,200 bp. Also, the intergenic regionof ycf1 and ndhF at the IRb and SSC border is larger(202 bp) as compared to other species. The only othergenome that has a larger border is Eucalyptus with218 bp.
The border regions of LSC-IRb, IRb- SSC, SSC-IRaand IRa-LSC keep changing in many species; though thegeneral arrangements of genes are consistent, theboundary regions are dynamic and changes are common.The expansion and contraction of the boundary regions are
not species and clade specific, and many changes areobserved in closely related species. Similarity in theboundary region between distantly related species is alsoobserved. The study of the IR junctions between LSC andSSC show remarkable changes between Jatropha and itsclosest relative Manihot (Fig. 4). Though the IR region inJatropha is larger as compared to Manihot the LSC-IRboundaries have shifted inwards from rps19 to rpl2. Inmost of the chloroplast genomes, the LSC-IR boundary iseither within the rps19 gene or beyond it; Jatropha is oneof the few cases where the LSC-IR boundary is within therpl2 gene. The two rpl2 genes differ in 13 nucleotides atthe end, and this resulted in change in three amino acids atthe end of the two genes. The SSC region in Jatropha isshorter as compared to Manihot. Interestingly, thedifference in the size of the SSC is similar to the differencein the size of the IR region between Jatropha andManihot. The IR region is 170 bp larger and SSC 398 bpshorter in Jatropha.
For the phylogenetic analysis, the matrix of 64 taxa(Jansen et al. 2007) has been used and 81 genes ofJatropha chloroplast has been added to it. The placementof Jatropha with Manihot has been supported with highBS in all the three trees made on the basis of MP, ML andME analyses. The Malpighiales group resolved well withhigh BS values. Jatropha and Manihot both belong to thefamily Euphorbiaceae. The phylogenetic analysis ofthe Euphorbiaceae family using molecular characters andthe chloroplast genes rbcL and trnL-F DNA sequencehave earlier been studied by Wurdack et al. 2005 andTokuoka 2007. Even in these studies, the inaperturatecrotonoids of which Jatropha is a member were sister toarticulated crotonoids of which Manihot is a member witha high bootstrap value of 100.
The accD gene that is an important chloroplastic gene ofthe lipid biosynthesis pathway did not show any significantchanges in Jatropha as compared to the other genomes.
166 bp
ndhF`rps 19
rpl 2ycf1`
ycf1
166 bp46 bpoverlap
1381 bp1381 bp
26954 bp 18250 bp
Ma
rpl 2
2 bp
trnH
rpl 2
rps 19 rpl 2
ycf1`
ycf1ndhF`
202 bp 2200 bp2200 bp
27124 bp 17852 bp
Ja13 bp
69 bp
trnH
13 bp
IRb IRaSSC
Fig. 4 Detailed view of the inverted repeat single-copy (IR/SC) border regions from Jatropha and Manihot with respect to the genes located at ornear the boundaries. Genes suffixed with an apostrophe represent partial genes. The figure is not to scale. Ma Manihot esculenta, Ja J. curcas
950 Tree Genetics & Genomes (2010) 6:941–952
Also, the major genes of the lipid biosynthesis pathway arenuclear-encoded; it could be their regulation and transportto chloroplast that may be regulating the oil biosynthesis inJatropha.
Conclusion
To summarise, this is the first report on chloroplast genomesequence of an oil-producing tree species of the Euphor-biaceae family. The chloroplast size and arrangement aresimilar to other land plant chloroplasts, with the exceptionof the loss of rps16 and infA genes. The phylogeneticanalyses by MP, ML and ME methods show that Jatrophaforms strong relationship with Manihot (100% BS).However, it also shares many similarities with Populus, atree species of the Eurosids clade, like loss of both the infAand the rps16 genes. The availability of the Jatrophachloroplast genome sequence can facilitate the design ofvectors for efficient transformation of Jatropha, resulting inbetter varieties.
Acknowledgements The Council of Scientific and Industrial Re-search, India, is acknowledged for supporting the research project.The authors wish to acknowledge the anonymous reviewers for theircritical reading and valuable suggestions. RT acknowledges DST forJC Bose Fellowship.
References
Basha SD, Sujatha M (2007) Inter and intra-population variability ofJatropha curcas (L.) characterized by RAPD and ISSR markersand development of population-specific SCAR markers. Euphy-tica 156:375–386
Bausher MG, Singh ND, Lee SB, Jansen RK, Daniell H (2006) Thecomplete chloroplast genome sequence of Citrus sinensis (L.)Osbeck var ‘ridge pineapple’: organization and phylogeneticrelationships to other angiosperms. BMC Plant Biol 6:21
Daniell H, Lee SB, Grevich J, Saski C, Quesada-Vargas T, Guda C,Tomkins J, Jansen RK (2006) Complete chloroplast genomesequences of Solanum bulbocastanum, Solanum lycopersicumand comparative analyses with other Solanaceae genomes. TheorAppl Genet 112:1503–1518
Daniell H, Wurdack KJ, Kanagaraj A, Lee SB, Saski C, Jansen RK(2008) The complete nucleotide sequence of the cassava(Manihot esculenta) chloroplast genome and the evolution ofatpF in Malpighiales: RNA editing and multiple losses of a groupII intron. Theor Appl Genet 116:723–737
Dixit R, Trivedi PK, Nath P, Sane PV (1999) Organization and post-transcriptional processing of the psb B operon from chloroplastsof Populus deltoides. Curr Genet 36:165–172
Jansen RK, Kaittanis C, Saski C, Lee SB, Tomkins J, Alverson AJ,Daniell H (2006) Phylogenetic analyses of Vitis (Vitaceae) basedon complete chloroplast genome sequences: effects of taxonsampling and phylogenetic methods on resolving relationshipsamong rosids. BMC Evol Biol 6:32
Jansen RK, Cai Z, Raubeson LA, Daniell H, Depamphilis CW,Leebens-Mack J, Muller KF, Guisinger-Bellian M, Haberle RC,Hansen AK, Chumley TW, Lee SB, Peery R, McNeal JR, KuehlJV, Boore JL (2007) Analysis of 81 genes from 64 plastidgenomes resolves relationships in angiosperms and identifiesgenome-scale evolutionary patterns. Proc Natl Acad Sci U S A104:19369–19374
Jones N, Miller JH (1991) Jatropha curcas a multipurpose species forproblematic sites. Land Resources Series 1
Kim KJ, Lee HL (2004) Complete chloroplast genome sequencesfrom Korean ginseng (Panax schinseng Nees) and comparativeanalysis of sequence evolution among 17 vascular plants. DNARes 11:247–261
King AJ, He W, Cuevas JA, Freudenberger M, Ramiaramanana D,Graham IA (2009) Potential of Jatropha curcas as a source ofrenewable oil and animal feed. J Exp Bot 60:2897–2905
Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J,Giegerich R (2001) REPuter: the manifold applications of repeatanalysis on a genomic scale. Nucleic Acids Res 29:4633–4642
Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, AntonescuC, Salzberg SL (2004) Versatile and open software for comparinglarge genomes. Genome Biol 5:R12
Lohse M, Drechsel O, Bock R (2007) OrganellarGenomeDRAW(OGDRAW): a tool for the easy generation of high-qualitycustom graphical maps of plastid and mitochondrial genomes.Curr Genet 52:267–274
Millen RS, Olmstead RG, Adams KL, Palmer JD, Lao NT, Heggie L,Kavanagh TA, Hibberd JM, Gray JC, Morden CW, Calie PJ,Jermiin LS, Wolfe KH (2001) Many parallel losses of infA fromchloroplast DNA during angiosperm evolution with multipleindependent transfers to the nucleus. Plant Cell 13:645–658
Nei M, Gojobori T (1986) Simple methods for estimating the numbersof synonymous and nonsynonymous nucleotide substitutions.Mol Biol Evol 3:418–426
Nei M, Kumar S (2000) Molecular evolution and phylogenetics.Oxford University Press, New York, p 128
Openshaw K (2000) A review of Jatropha curcas: an oil plant ofunfulfilled promise. Biomass Bioenergy 19:1–15
Page RD (1996) TreeView: an application to display phylogenetictrees on personal computers. Comput Appl Biosci 12:357–358
Raubeson LA, Jansen RK (2005) In diversity and evolution of plants-genotypic and phenotypic variation in higher plants. In: WallingfordHH (ed) Chloroplast genomes of plants. CABI, Wallingford,pp 45–68
Raubeson LA, Peery R, Chumley TW, Dziubek C, Fourcade HM,Boore JL, Jansen RK (2007) Comparative chloroplast genomics:analyses including new sequences from the angiosperms Nupharadvena and Ranunculus macranthus. BMC Genomics 8:174
Ravi V, Khurana JP, Tyagi AK, Khurana P (2006) The chloroplastgenome of mulberry: complete nucleotide sequence, geneorganization and comparative analysis. Tree Genet Genomes3:49–59
Ruhlman T, Lee SB, Jansen RK, Hostetler JB, Tallon LJ, Town CD,Daniell H (2006) Complete plastid genome sequence of Daucuscarota: implications for biotechnology and phylogeny of angio-sperms. BMC Genomics 7:222
Saitou N, Nei M (1987) The neighbor-joining method: a new methodfor reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Shimada H, Sugiura M (1991) Fine structural features of thechloroplast genome: comparison of the sequenced chloroplastgenomes. Nucleic Acids Res 19:983–995
Sudheer Pamidiamarri DV, Pandya N, Reddy MP, Radhakrishnan T(2009) Comparative study of interspecific genetic divergence andphylogenic analysis of genus Jatropha by RAPD and AFLP:genetic divergence and phylogenic analysis of genus Jatropha.Mol Biol Rep 36:901–907
Tree Genetics & Genomes (2010) 6:941–952 951
Sujatha M, Reddy TP, Mahasi MJ (2008) Role of biotechnologicalinterventions in the improvement of castor (Ricinus communis L.)and Jatropha curcas L. Biotechnol Adv 26:424–435
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: molecularevolutionary genetics analysis (MEGA) software version 4.0.Mol Biol Evol 24:1596–1599
Tanaka M, Obokata J, Chunwongse J, Shinozaki K, Sugiura M (1987)Rapid splicing and stepwise processing of a transcript from thepsbB operon in tobacco chloroplasts: determination of the intronsites in petB and petD. Mol Gen Genet 209:427–431
Tokuoka T (2007) Molecular phylogenetic analysis of Euphorbiaceaesensu stricto based on plastid and nuclear DNA sequences andovule and seed character evolution. J Plant Res 120:511–522
Tsudzuki T,Wakasugi T, SugiuraM (2001) Comparative analysis of RNAediting sites in higher plant chloroplasts. J Mol Evol 53:327–332
Ueda M, Nishikawa T, Fujimoto M, Takanashi H, Arimura S,Tsutsumi N, Kadowaki K (2008) Substitution of the gene forchloroplast RPS16 was assisted by generation of a dual targetingsignal. Mol Biol Evol 25:1566–1575
Wurdack KJ, Hoffmann P, Chase MW (2005) Molecular phylogeneticanalysis of uniovulate Euphorbiaceae (Euphorbiaceae sensustricto) using plastid RBCL and TRNL-F DNA sequences. AmJ Bot 92:1397–1420
Zwickl DJ (2006) Genetic algorithm approaches for the phyloge-netic analysis of large biological sequence datasets under themaximum likelihood criterion. The University of Texas,Austin