Contents lists available at SciVerse ScienceDirect
Colloids and Surfaces A: Physicochemical andEngineering Aspects
jo ur nal ho me page: www.elsev ier .com/ locate /co lsur fa
-Lactoglobulin, gum arabic, and xanthan gum for emulsifying sweetlmond oil: Formulation and stabilization mechanisms ofharmaceutical emulsions
léonore Bouyera,b, Ghozlene Mekhloufia,b,∗, Nicolas Huanga,b,éronique Rosilioa,b, Florence Agnelya,b
Univ. Paris Sud, Faculté de Pharmacie, 5 rue Jean-Baptiste Clément, 92296 Châtenay-Malabry Cedex, FranceCNRS, UMR 8612, Institut Galien Paris-Sud, 5 rue Jean-Baptiste Clément, 92296 Châtenay-Malabry Cedex, France
i g h l i g h t s
Replace synthetic surfactants bybiopolymers in emulsions forpharmaceutical use.Formulate and characterize O/Wemulsions stabilized by �-lg, GA, XG,�-lg:GA, and �-lg:XG.Interfacial tension and rheologicalmeasurements to study stabilizationmechanisms.Interfacial adsorption for �-lg andGA. Thickening of the continuousphase by XG.�-lg:GA: interfacial bi-layer. �-lg:XG: �-lg adsorption and XGthickening.
g r a p h i c a l a b s t r a c t
a r t i c l e i n f o
rticle history:eceived 6 February 2013eceived in revised form 29 April 2013ccepted 30 April 2013vailable online 9 May 2013
eywords:-Lactoglobulin
a b s t r a c t
Emulsions are thermodynamically unstable systems generally stabilized by small surfactant molecules.To answer sustainable development concerns, it is possible to replace the latter by natural emulsifierssuch as proteins and polysaccharides to formulate emulsions for the pharmaceutical field. In this work,we describe oil-in-water emulsions stabilized by natural biopolymers. In a first approach, we preparedemulsions with various binary mixtures of �-lactoglobulin (�-lg), gum arabic (GA) and xanthan gum (XG),and assessed their stability over a 6-month period by zeta potential and granulometry measurementsin combination with Turbiscan and optical microscopy observations. The efficacy of the biopolymers to
um arabicanthan gummulsiontabilitynterface
stabilize emulsions was in the order GA < �-lg:GA < �-lg < XG = �-lg:XG. In a second approach, we focusedon the determination of the stabilization mechanisms induced by these biopolymers, by interfacial ten-sion and rheological measurements. Different stabilization mechanisms could be highlighted: interfacialadsorption for �-lg and GA winterfacial adsorption, formati�-lg interfacial adsorption and
In recent years, pharmaceutical and cosmetic industries haveshown increasing interest regarding the use of “clean labelled”ingredients and sustainable development [1,2]. Biopolymers are
urrently used in the food industry as texturizing and stabiliz-ng agents [3–6], and could replace synthetic surfactants in theormulation of pharmaceutical emulsions [7]. Indeed, these syn-hetic surfactants may cause acute toxic symptoms in animals andumans and have an environmental impact [8–10]. Thus, replac-
ng them by much safer molecules would be a major opening. Iniopolymer-stabilized emulsions, proteins generally act as maintabilizers and polysaccharides contribute through their thickeningnd steric stabilizing properties [11]. Most polysaccharides formn extended network in the continuous phase that increases itsiscosity [12], even lead to a gel [13,14], slowing down dropletsovements and encounters. However, some polysaccharides are
lso able to adsorb at droplets interface by their hydrophobicesidues, decreasing the interfacial tension and forming a stericarrier against coalescence [15]. Combining in a same systemhe properties of proteins (fast adsorption, significant decrease innterfacial tension) and polysaccharides (steric repulsion or viscos-ty enhancement) under appropriate conditions (concentrations,rotein-to-polysaccharide ratio, pH, ionic strength, temperature)ould be of great interest for improving emulsion stability [16–19].ctually, proteins and polysaccharides both partake in creatingovel emulsions with improved stability and functionality. Whenoth kinds of biopolymers are associated in appropriate physico-hemical conditions so that they carry opposite charges, themulsion stability is provided by the resulting surface active pro-ein:polysaccharide complex. The protein can first adsorb at thenterface, and the polysaccharide forms an interfacial complex witht through attractive electrostatic interactions. It is then possible toreate multi-layered “membranes” around oil droplets by adsorb-ng alternative layers of oppositely charged biopolymers [19]. Theormation of a protein:polysaccharide complex has been reportedo enhance thermal stability and resistance of emulsions to externalreatments (i.e. thermal processing, chilling and freezing) [19–21].
oreover, emulsions can be stabilized by protein and polysaccha-ide without development of any attractive interactions betweenhem: the protein adsorbs at the interface, whereas the polysac-haride increases the viscosity of the continuous phase. The fieldf biopolymer-stabilized emulsions has recently been reviewed7,12,16] and the interest of such systems for pharmaceutical appli-ations has been demonstrated [7].
In the present study, three biopolymers have been chosen totabilize oil-in-water emulsions: �-lactoglobulin (�-lg), the majorhey protein, gum arabic (GA), an exudate from Acacia trees,
nd xanthan gum (XG), produced by the bacterium Xanthomonasampestris.
�-lg is a globular protein that contains 162 amino acid residues,ne thiol group and two disulphide bonds. The molar mass of aonomer is 18.1 × 103 g/mol [22] and the isoelectric point (pI)
s about 5.2. �-lg is currently used with success in food emul-ions [23,24]. It rapidly adsorbs at the oil–water interface, partiallynfolds in order to expose its hydrophobic residues towards the
ipophilic phase, and lowers the interfacial tension [25,26]. It formsnterfacial elastic films that prevent coalescence [27,28]. Moreover,he remaining protein residues in the aqueous phase provide sterictabilization that protects dispersed droplets against coalescence,reaming, and flocculation [29,30].
GA is a hetero-polysaccharide containing both protein andolysaccharide residues. It consists of three fractions: arabinogalac-an (AG) (80–90 wt% of the total gum), glycoprotein (GP) (2–4 wt%f the total gum), and arabinogalactan-protein complex (AGP)10–20 wt% of the total gum) [31]. The glucuronic acid residues ints chemical structure confer a net negative charge to the gum above
H 2.0. Molar mass of GA varies from 3.0 × 105 to 5.8 × 105 g/molepending on its origin and age [32]. The AGP fraction has beenonsidered to be mostly responsible for the emulsifying propertiesf the gum [33–36]. Indeed, the AGP has a “wattle blossom” type
icochem. Eng. Aspects 433 (2013) 77– 87
structure with polysaccharidic blocks of molecular mass ∼ 4 × 104
linked to a polypeptide chain consisting of ∼250 amino acidsthrough serine and hydro linkages [37]. This structure confers anamphiphilic character to the gum and allows its adsorption at theoil–water or air–water interface [32,38]. The proteinaceous com-ponents of the gum would embed in the air or in the oil phase, whilethe carbohydrate ones would extend out from the surface intothe aqueous phase [35,39]. The adsorbed gum arabic at oil–waterinterface can prevent droplet flocculation and coalescence throughboth electrostatic and steric repulsive forces [37].
XG is an anionic polysaccharide, which molar mass varies from1.5 × 106 to 5 × 106 g/mol depending on fermentation conditionsand content in acetate and pyruvate residues, influencing solutionviscosity [40]. Circular dichroism measurements showed evidenceof two characteristic conformations (random and ordered) of XG,regardless of the temperature and concentrations of salt and poly-mer [41]. The stiff polymer chain may exist as a single, double ortriple helix that interacts with another molecule to form a complex,loosely bound network in solution [42,43]. This particular structuregives the gum its unusual rheological characteristics in solutions:high low-shear viscosity and strong shear-thinning character. XG isthus a good emulsion stabilizer, enhancing droplet stability againstflocculation and creaming [44].
In a recent study [27], our group has formulated �-lg, GA, and �-lg:GA stabilized emulsions. Interfacial measurements revealed thatfollowing protein adsorption at the oil–water interface, GA elec-trostatically bound to it, thus forming a second layer around theglobules. In the continuity of this work, the present study aimed atevaluating the long term stability of these systems and comparingit to that of �-lg:XG stabilized emulsions. Thus, in the current study,several emulsions were formulated and stabilized by �-lg, GA, �-lg:GA, and �-lg:XG. �-lg and GA at a 2:1 ratio were associated atpH 4.2 to allow formation of a complex by attractive electrostaticinteractions [45,46]. Some studies have shown that the 2:1 �-lg:GAratio at pH 4.2 induced a maximum interaction between the twobiopolymers [45,46]. Conversely, �-lg and XG were mixed at pH 7in a 1:1 ratio, where both biopolymers displayed an anionic charge,in order to avoid formation of a complex and ensure that XG wouldonly act as a thickener in the continuous phase. The assessment ofemulsions stability was conducted over a 6 months period, com-bining different techniques such as Turbiscan, zeta potential, anddroplet size measurements. Moreover, the emulsion stabilizationmechanisms by these biopolymers were analyzed by rheology andby interfacial and surface tension measurements. The influence ofeach emulsifier on the composition of the interface was thus eval-uated and compared. The knowledge gained in the present studywill be usefull from a physico-chemical standpoint and for furtherformulation development.
2. Materials and methods
2.1. Materials
�-lg (batch JE 002-8-992) was supplied by Davisco Foods Inter-national, Inc. (USA). The powder composition was 89.8 (w/w%)protein, 8.8 (w/w%) moisture, and 1.4 (w/w%) ash [45]. GA(INSTANTGUM AA, with a molar mass of about 350,000 g/mol asreported by the supplier) was a gift from CNI Company (Rouen,France). Its composition included 10 (w/w%) moisture, 4 (w/w%)ash, and a 2.5 (w/w%) protein content (as determined by Kjeldahlanalysis with N × 6.66). XG (SatiaxaneTM CX 910, lot 20060134)
was generously given by Degussa (Paris, France). As reported bythe supplier, the powder contained 7 (w/w%) proteins, 11 (w/w%)moisture, and 9 (w/w%) ash. Sweet almond oil (batch 07120145\A)was provided by Cooper (Melun, France) and sodium azide (batch
305.2) by Roth (Lauterbourg, France). The materials were noturified, according to their usual industrial use. Sodium hydrox-
de, hydrochloric acid, and citric acid were analytical grades fromWR (Fontenay-sous-Bois, France). Water used in all experimentsas purified using a Millipore Synergy 185 apparatus coupled to aiOs5TM, with a resistivity of 18.2 M� cm.
.2. Preparation of biopolymer solutions
Biopolymer solutions were prepared at total final concentra-ions of 1 and 2.5 (w/w%) for �-lg and GA, and 0.1 and 1 (w/w%)or XG. The powders were dissolved in MilliQ water under gentletirring at 20 ± 1 ◦C for at least 2 h. XG solutions were heated to0 ◦C immediately after powder dispersion for 30 min in order tollow a better solubilization of the gum. All biopolymer solutionsere then allowed to hydrate overnight at 4 ± 1 ◦C. Sodium azide
0.05 (w/w%)) was added as an antimicrobial agent. The pH of �-lgolutions was adjusted to 4.8 (lowest �-lg solubility pH [45]) using.1 or 1 N HCl solutions. �-lg solutions were then centrifuged at0,000 × g, 20 ± 1 ◦C for 30 min to remove insoluble particles and-lg aggregates. This step induced a 10% �-lg loss [45] that was
aken into account when preparing the protein solutions. The finalH of �-lg solutions was then adjusted to either 4.2 or 7.0 using.1 or 1 N HCl solutions or 1 N NaOH solution. The pH of GA and XGolutions was adjusted to 4.2 and 7.0, respectively.
.3. Preparation of oil-in-water emulsion
Emulsions were prepared by homogenizing 30 (w/w%) sweetlmond oil with aqueous biopolymer solutions. Five differentmulsifier compositions were studied: �-lg, GA, �-lg:GA, XG, and-lg:XG (Fig. 1). The total biopolymer concentration was first set to% and when emulsions were not stable, it was increased to 2.5%.
�-lg and GA stabilized emulsions were formulated by adding theil to the biopolymer solution in a rotor-stator homogenizer (Poly-ron PT-MR 3100, Kinematica AG, Bioblock Scientific) for 2 min at5,000 rpm. Emulsions were then passed through a two-stage high-ressure valve homogenizer (APV Invensys) for 5 min and with twouccessive pressure steps, 500 and 50 bars (Fig. 1, ).
Mixed �-lg:GA stabilized emulsions were formulated at a pro-ein:polysaccharide ratio of 2:1 by adding a GA solution to areviously formed �-lg stabilized emulsion at pH 4.2, by simpleand reversal shaking to minimize system destructuring (Fig. 1, ).
ig. 1. Schematic depiction of the protocols for emulsions preparation with �-lgr GA, mixed �-lg:GA, XG, and mixed �-lg:XG solutions. rs, rotor-stator homog-nizer; hp, high pressure homogenizer; H, hand reversal shaking; thermometericture, heating; T, Turbotest Rayneri paddle.
icochem. Eng. Aspects 433 (2013) 77– 87 79
The preparation of XG emulsions consisted in heating the oilyphase and the aqueous XG solution to 50 ◦C in order to lower theirviscosities. The oil phase was then added to the continuous phaseusing a Turbotest Rayneri paddle and left for 45 min at 2600 rpm. XGstabilized emulsions were finally passed in a rotor-stator homoge-nizer for 5 min at 15,000 rpm (Fig. 1, ).
�-lg:XG stabilized emulsions were formulated with a 1:1 �-lg:XG ratio using the same protocol as XG stabilized emulsions, withthe difference that the oily phase was first dispersed into the �-lgsolution with the rotor-stator homogenizer. XG solution was thenadded to the �-lg stabilized emulsions using a Turbotest Rayneripaddle for 2 min at 1000 rpm (Fig. 1, ).
It is important to note that the formulation protocols were notidentical for all emulsions. We first tried to formulate all the emul-sions without using any high-pressure valve homogenizer to havethe lowest possible energy expense in the formulation process.However, �-lg, GA, and �-lg:GA stabilized emulsions phase sep-arated immediately, probably due to the large sizes of the formeddroplets and to the low viscosities of the continuous phases (resultsnot shown). These results explain the use of a high-pressure valvehomogenizer to prepare these emulsions, and thus the differencesin the formulation protocols when using XG. All these emulsionswere formulated to study their stability over a 6-month period.
2.4. Assessment of emulsion stability
Emulsions were characterized only when stable for at least 24 h.All measurements were performed on the first day of stability andregularly over a 6-month period (except for zeta potential measure-ments) as long as no phase separation was visually noticed. Duringthis period, emulsions were stored at room temperature and awayfrom light.
2.4.1. Turbiscan measurementsThe stability of emulsions was evaluated using a Turbiscan Clas-
sic MA 2000 (Formulaction®, L’Union, France). This technique isbased on multiple light scattering, using a pulsed near infraredlight source (� = 850 nm). The apparatus is composed of a readinghead and two synchronous detectors that scan by up and downmovements along a glass cylindrical cell filled with the sample. Thetransmission detector measures the light flux transmitted (at theangle of 0◦ from the incident beam) through the emulsion, and thebackscattering detector receives the light backscattered (at 135◦
from the incident beam) by the emulsion. Retrodiffusion and trans-mission intensities versus sample height and time are obtained,thus allowing detection of flocculation and coalescence processeseven at a very early stage (invisible to the eye) [47,48]. Turbiscanmeasurements were performed over a 6-month period, daily dur-ing the 15 days after the formulation, and then monthly (days 30,60, 90, 120, 150, and 180).
2.4.2. Zeta potential measurementsThe �-potential was determined by measuring the direction
and velocity of droplet movement in a well-defined electric field.The measurements were performed using a laser doppler micro-electrophoresis (Zetasizer Nano ZS 90, Malvern Instruments, Orsay,France) equipped with a 633 nm helium-neon laser. Scattered lightwas analyzed at an angle of 90◦. The �-potential was calculatedusing the Smoluchowski model. Emulsions were diluted to 1/50with MilliQ water prior to analysis. The measurements were con-ducted at 25 ◦C and in triplicate on the first day of stability. Resultsare mean �-potential values of oil droplets.
2.4.3. Droplet size determinationsEmulsions droplet size was measured by laser diffraction. The
mean volume diameter, d43, of emulsions droplet was determined
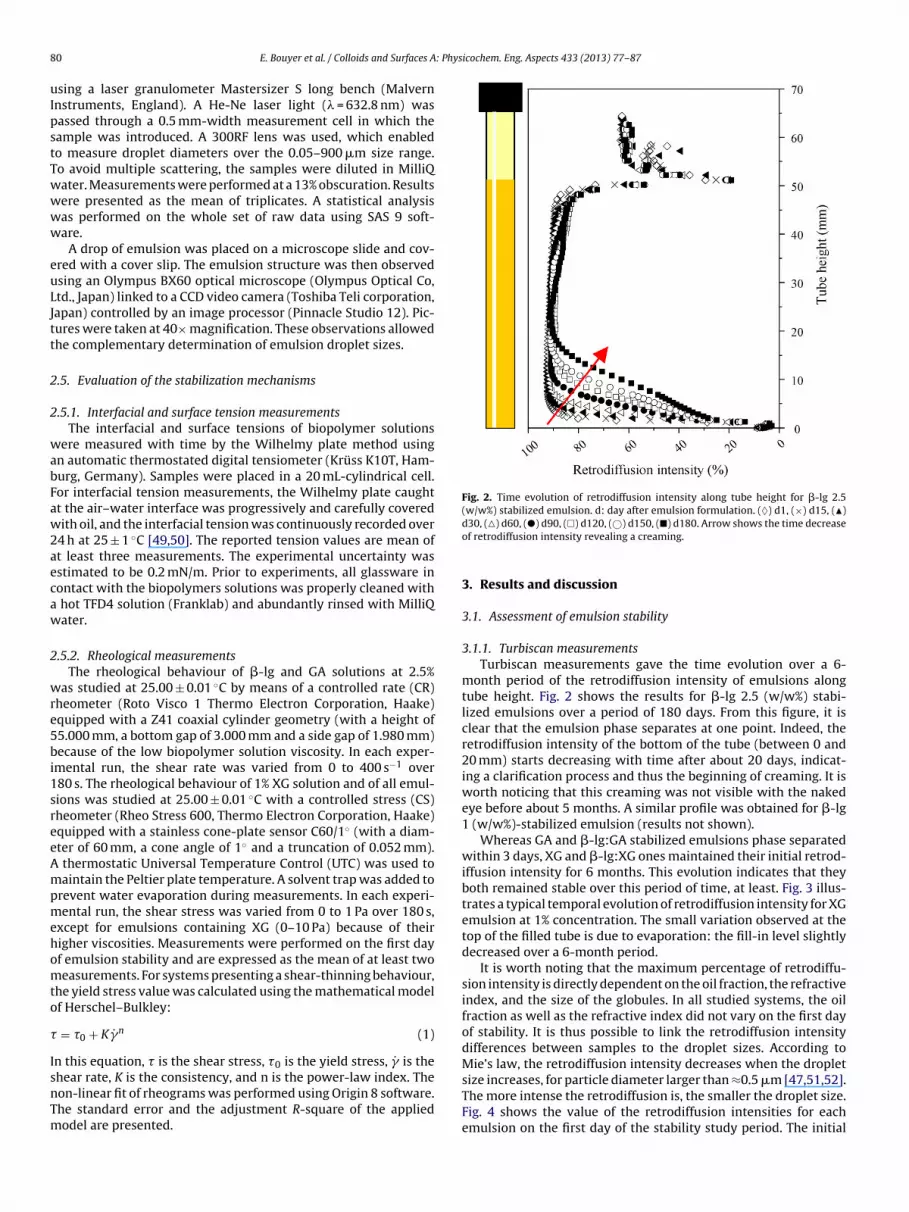
Fig. 2. Time evolution of retrodiffusion intensity along tube height for �-lg 2.5(w/w%) stabilized emulsion. d: day after emulsion formulation. (♦) d1, (×) d15, (�)
0 E. Bouyer et al. / Colloids and Surfaces A
sing a laser granulometer Mastersizer S long bench (Malvernnstruments, England). A He-Ne laser light (� = 632.8 nm) wasassed through a 0.5 mm-width measurement cell in which theample was introduced. A 300RF lens was used, which enabledo measure droplet diameters over the 0.05–900 �m size range.o avoid multiple scattering, the samples were diluted in MilliQater. Measurements were performed at a 13% obscuration. Resultsere presented as the mean of triplicates. A statistical analysisas performed on the whole set of raw data using SAS 9 soft-are.
A drop of emulsion was placed on a microscope slide and cov-red with a cover slip. The emulsion structure was then observedsing an Olympus BX60 optical microscope (Olympus Optical Co,td., Japan) linked to a CCD video camera (Toshiba Teli corporation,apan) controlled by an image processor (Pinnacle Studio 12). Pic-ures were taken at 40× magnification. These observations allowedhe complementary determination of emulsion droplet sizes.
.5. Evaluation of the stabilization mechanisms
.5.1. Interfacial and surface tension measurementsThe interfacial and surface tensions of biopolymer solutions
ere measured with time by the Wilhelmy plate method usingn automatic thermostated digital tensiometer (Krüss K10T, Ham-urg, Germany). Samples were placed in a 20 mL-cylindrical cell.or interfacial tension measurements, the Wilhelmy plate caughtt the air–water interface was progressively and carefully coveredith oil, and the interfacial tension was continuously recorded over
4 h at 25 ± 1 ◦C [49,50]. The reported tension values are mean oft least three measurements. The experimental uncertainty wasstimated to be 0.2 mN/m. Prior to experiments, all glassware inontact with the biopolymers solutions was properly cleaned with
hot TFD4 solution (Franklab) and abundantly rinsed with MilliQater.
.5.2. Rheological measurementsThe rheological behaviour of �-lg and GA solutions at 2.5%
as studied at 25.00 ± 0.01 ◦C by means of a controlled rate (CR)heometer (Roto Visco 1 Thermo Electron Corporation, Haake)quipped with a Z41 coaxial cylinder geometry (with a height of5.000 mm, a bottom gap of 3.000 mm and a side gap of 1.980 mm)ecause of the low biopolymer solution viscosity. In each exper-
mental run, the shear rate was varied from 0 to 400 s−1 over80 s. The rheological behaviour of 1% XG solution and of all emul-ions was studied at 25.00 ± 0.01 ◦C with a controlled stress (CS)heometer (Rheo Stress 600, Thermo Electron Corporation, Haake)quipped with a stainless cone-plate sensor C60/1◦ (with a diam-ter of 60 mm, a cone angle of 1◦ and a truncation of 0.052 mm).
thermostatic Universal Temperature Control (UTC) was used toaintain the Peltier plate temperature. A solvent trap was added to
revent water evaporation during measurements. In each experi-ental run, the shear stress was varied from 0 to 1 Pa over 180 s,
xcept for emulsions containing XG (0–10 Pa) because of theirigher viscosities. Measurements were performed on the first dayf emulsion stability and are expressed as the mean of at least twoeasurements. For systems presenting a shear-thinning behaviour,
he yield stress value was calculated using the mathematical modelf Herschel–Bulkley:
= �0 + K �n (1)
n this equation, � is the shear stress, �0 is the yield stress, � is the
hear rate, K is the consistency, and n is the power-law index. Theon-linear fit of rheograms was performed using Origin 8 software.he standard error and the adjustment R-square of the appliedodel are presented.
3.1.1. Turbiscan measurementsTurbiscan measurements gave the time evolution over a 6-
month period of the retrodiffusion intensity of emulsions alongtube height. Fig. 2 shows the results for �-lg 2.5 (w/w%) stabi-lized emulsions over a period of 180 days. From this figure, it isclear that the emulsion phase separates at one point. Indeed, theretrodiffusion intensity of the bottom of the tube (between 0 and20 mm) starts decreasing with time after about 20 days, indicat-ing a clarification process and thus the beginning of creaming. It isworth noticing that this creaming was not visible with the nakedeye before about 5 months. A similar profile was obtained for �-lg1 (w/w%)-stabilized emulsion (results not shown).
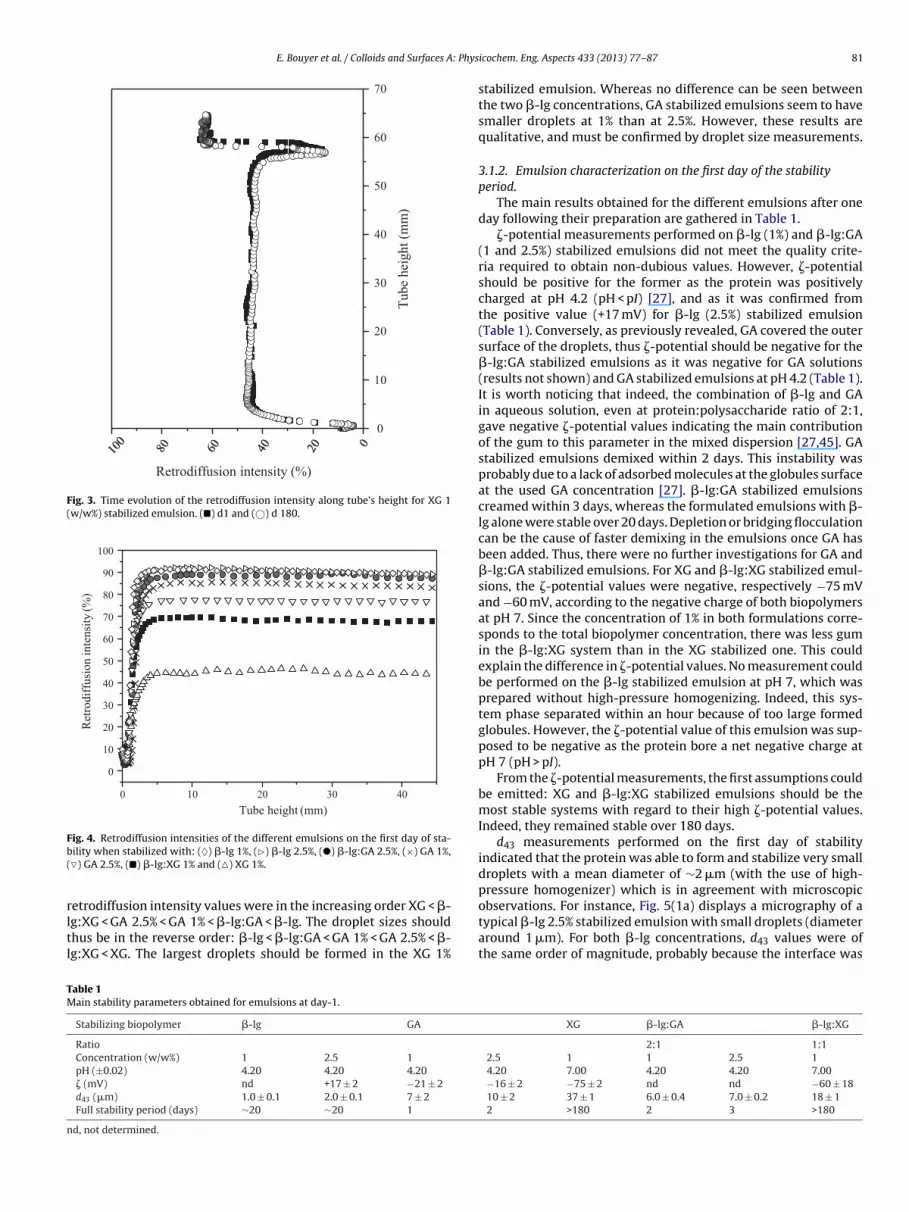
Whereas GA and �-lg:GA stabilized emulsions phase separatedwithin 3 days, XG and �-lg:XG ones maintained their initial retrod-iffusion intensity for 6 months. This evolution indicates that theyboth remained stable over this period of time, at least. Fig. 3 illus-trates a typical temporal evolution of retrodiffusion intensity for XGemulsion at 1% concentration. The small variation observed at thetop of the filled tube is due to evaporation: the fill-in level slightlydecreased over a 6-month period.
It is worth noting that the maximum percentage of retrodiffu-sion intensity is directly dependent on the oil fraction, the refractiveindex, and the size of the globules. In all studied systems, the oilfraction as well as the refractive index did not vary on the first dayof stability. It is thus possible to link the retrodiffusion intensitydifferences between samples to the droplet sizes. According toMie’s law, the retrodiffusion intensity decreases when the dropletsize increases, for particle diameter larger than ≈0.5 �m [47,51,52].
The more intense the retrodiffusion is, the smaller the droplet size.Fig. 4 shows the value of the retrodiffusion intensities for eachemulsion on the first day of the stability study period. The initial
observations. For instance, Fig. 5(1a) displays a micrography of a
TM
n
ig. 4. Retrodiffusion intensities of the different emulsions on the first day of sta-ility when stabilized with: (♦) �-lg 1%, (�) �-lg 2.5%, (�) �-lg:GA 2.5%, (×) GA 1%,�) GA 2.5%, (�) �-lg:XG 1% and (�) XG 1%.
etrodiffusion intensity values were in the increasing order XG < �-
g:XG < GA 2.5% < GA 1% < �-lg:GA < �-lg. The droplet sizes shouldhus be in the reverse order: �-lg < �-lg:GA < GA 1% < GA 2.5% < �-g:XG < XG. The largest droplets should be formed in the XG 1%
able 1ain stability parameters obtained for emulsions at day-1.
Stabilizing biopolymer �-lg GA
Ratio
Concentration (w/w%) 1 2.5 1
pH (±0.02) 4.20 4.20 4.20
� (mV) nd +17 ± 2 −21 ± 2
d43 (�m) 1.0 ± 0.1 2.0 ± 0.1 7 ± 2
Full stability period (days) ∼20 ∼20 1
d, not determined.
icochem. Eng. Aspects 433 (2013) 77– 87 81
stabilized emulsion. Whereas no difference can be seen betweenthe two �-lg concentrations, GA stabilized emulsions seem to havesmaller droplets at 1% than at 2.5%. However, these results arequalitative, and must be confirmed by droplet size measurements.
3.1.2. Emulsion characterization on the first day of the stabilityperiod.
The main results obtained for the different emulsions after oneday following their preparation are gathered in Table 1.
�-potential measurements performed on �-lg (1%) and �-lg:GA(1 and 2.5%) stabilized emulsions did not meet the quality crite-ria required to obtain non-dubious values. However, �-potentialshould be positive for the former as the protein was positivelycharged at pH 4.2 (pH < pI) [27], and as it was confirmed fromthe positive value (+17 mV) for �-lg (2.5%) stabilized emulsion(Table 1). Conversely, as previously revealed, GA covered the outersurface of the droplets, thus �-potential should be negative for the�-lg:GA stabilized emulsions as it was negative for GA solutions(results not shown) and GA stabilized emulsions at pH 4.2 (Table 1).It is worth noticing that indeed, the combination of �-lg and GAin aqueous solution, even at protein:polysaccharide ratio of 2:1,gave negative �-potential values indicating the main contributionof the gum to this parameter in the mixed dispersion [27,45]. GAstabilized emulsions demixed within 2 days. This instability wasprobably due to a lack of adsorbed molecules at the globules surfaceat the used GA concentration [27]. �-lg:GA stabilized emulsionscreamed within 3 days, whereas the formulated emulsions with �-lg alone were stable over 20 days. Depletion or bridging flocculationcan be the cause of faster demixing in the emulsions once GA hasbeen added. Thus, there were no further investigations for GA and�-lg:GA stabilized emulsions. For XG and �-lg:XG stabilized emul-sions, the �-potential values were negative, respectively −75 mVand −60 mV, according to the negative charge of both biopolymersat pH 7. Since the concentration of 1% in both formulations corre-sponds to the total biopolymer concentration, there was less gumin the �-lg:XG system than in the XG stabilized one. This couldexplain the difference in �-potential values. No measurement couldbe performed on the �-lg stabilized emulsion at pH 7, which wasprepared without high-pressure homogenizing. Indeed, this sys-tem phase separated within an hour because of too large formedglobules. However, the �-potential value of this emulsion was sup-posed to be negative as the protein bore a net negative charge atpH 7 (pH > pI).
From the �-potential measurements, the first assumptions couldbe emitted: XG and �-lg:XG stabilized emulsions should be themost stable systems with regard to their high �-potential values.Indeed, they remained stable over 180 days.
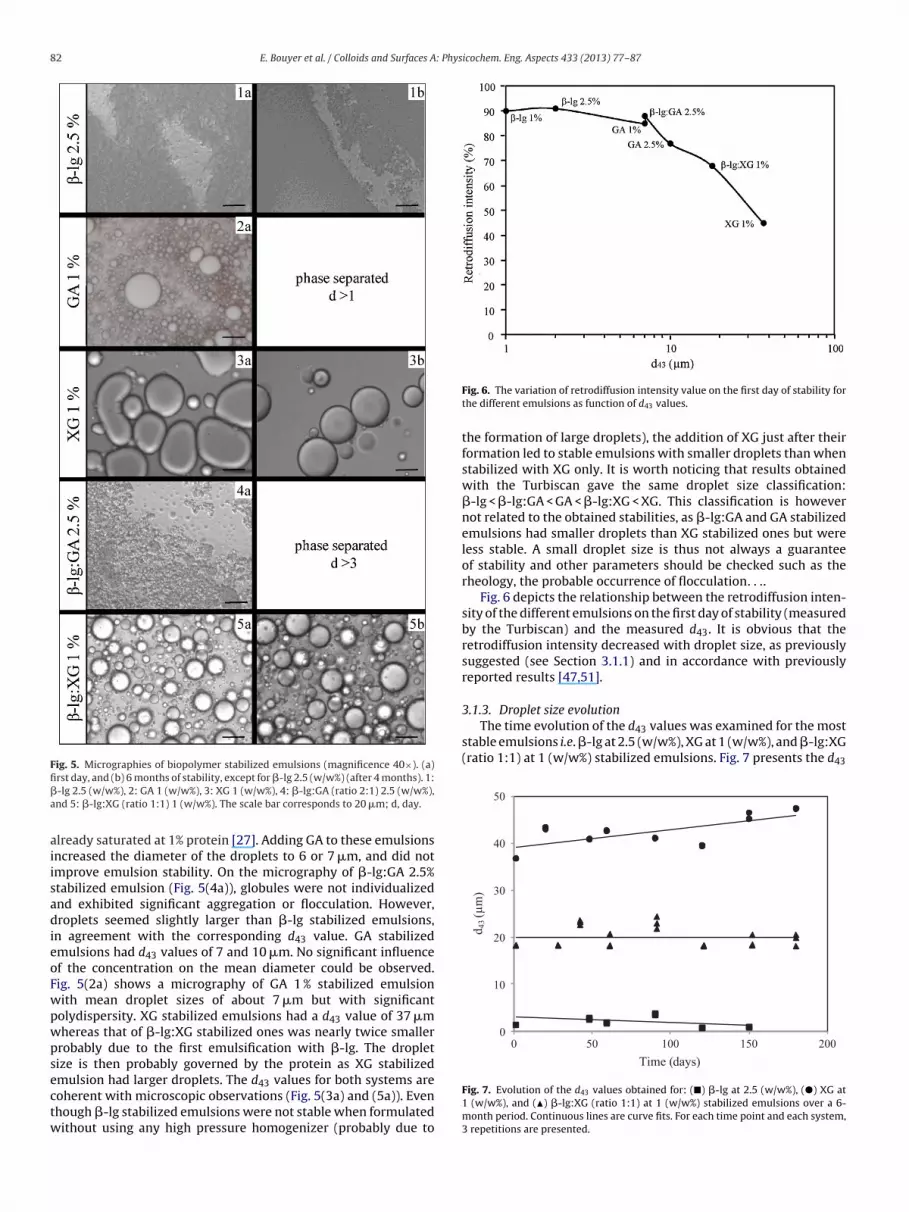
d43 measurements performed on the first day of stabilityindicated that the protein was able to form and stabilize very smalldroplets with a mean diameter of ∼2 �m (with the use of high-pressure homogenizer) which is in agreement with microscopic
typical �-lg 2.5% stabilized emulsion with small droplets (diameteraround 1 �m). For both �-lg concentrations, d43 values were ofthe same order of magnitude, probably because the interface was
82 E. Bouyer et al. / Colloids and Surfaces A: Physicochem. Eng. Aspects 433 (2013) 77– 87
Fig. 5. Micrographies of biopolymer stabilized emulsions (magnificence 40×). (a)fi�a
aiisadieoFwpwpsectw
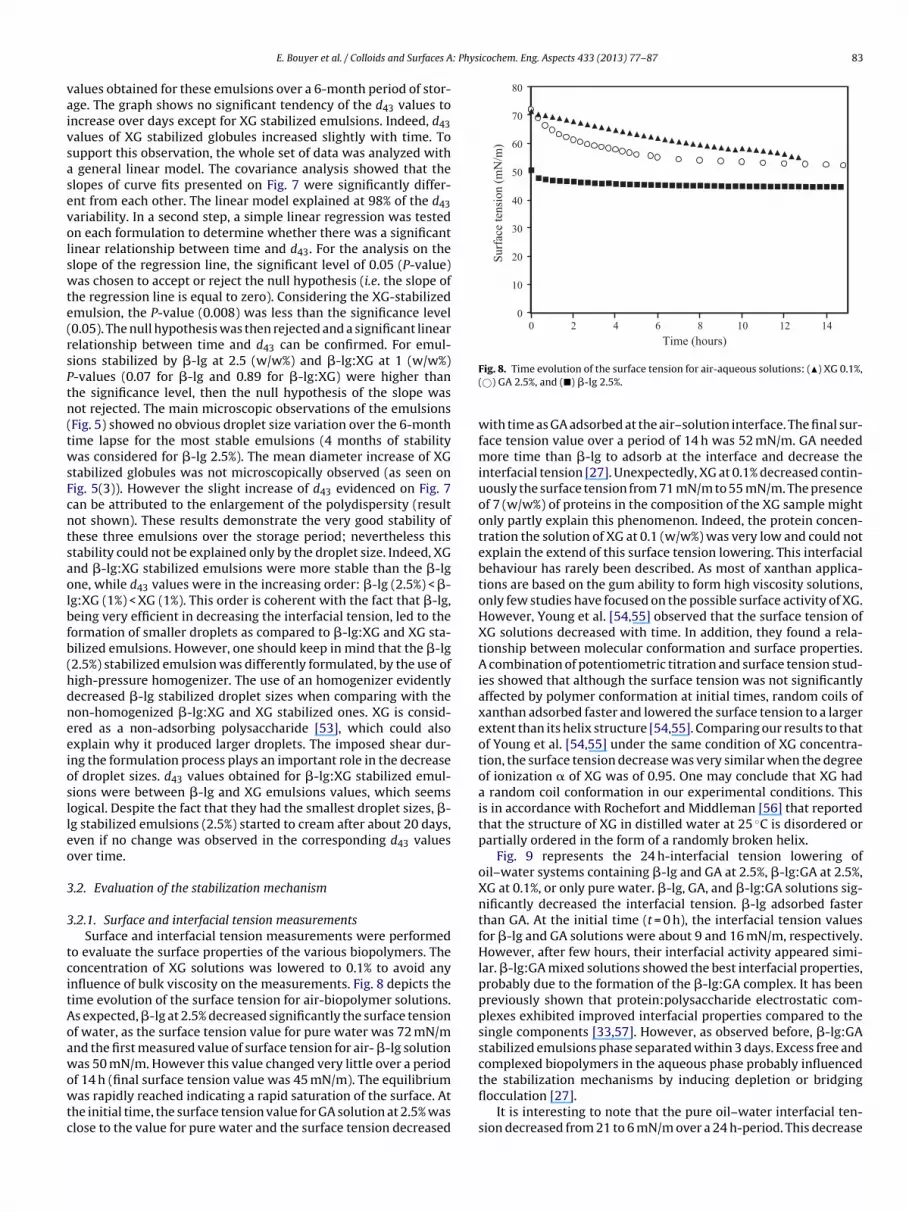
3.1.3. Droplet size evolutionThe time evolution of the d43 values was examined for the most
stable emulsions i.e. �-lg at 2.5 (w/w%), XG at 1 (w/w%), and �-lg:XG(ratio 1:1) at 1 (w/w%) stabilized emulsions. Fig. 7 presents the d43
0
10
20
30
40
50
0 50 100 150 200
d43
(µm
)
Time (days )
rst day, and (b) 6 months of stability, except for �-lg 2.5 (w/w%) (after 4 months). 1:-lg 2.5 (w/w%), 2: GA 1 (w/w%), 3: XG 1 (w/w%), 4: �-lg:GA (ratio 2:1) 2.5 (w/w%),nd 5: �-lg:XG (ratio 1:1) 1 (w/w%). The scale bar corresponds to 20 �m; d, day.
lready saturated at 1% protein [27]. Adding GA to these emulsionsncreased the diameter of the droplets to 6 or 7 �m, and did notmprove emulsion stability. On the micrography of �-lg:GA 2.5%tabilized emulsion (Fig. 5(4a)), globules were not individualizednd exhibited significant aggregation or flocculation. However,roplets seemed slightly larger than �-lg stabilized emulsions,
n agreement with the corresponding d43 value. GA stabilizedmulsions had d43 values of 7 and 10 �m. No significant influencef the concentration on the mean diameter could be observed.ig. 5(2a) shows a micrography of GA 1 % stabilized emulsionith mean droplet sizes of about 7 �m but with significantolydispersity. XG stabilized emulsions had a d43 value of 37 �mhereas that of �-lg:XG stabilized ones was nearly twice smallerrobably due to the first emulsification with �-lg. The dropletize is then probably governed by the protein as XG stabilized
mulsion had larger droplets. The d43 values for both systems areoherent with microscopic observations (Fig. 5(3a) and (5a)). Evenhough �-lg stabilized emulsions were not stable when formulatedithout using any high pressure homogenizer (probably due to
Fig. 6. The variation of retrodiffusion intensity value on the first day of stability forthe different emulsions as function of d43 values.
the formation of large droplets), the addition of XG just after theirformation led to stable emulsions with smaller droplets than whenstabilized with XG only. It is worth noticing that results obtainedwith the Turbiscan gave the same droplet size classification:�-lg < �-lg:GA < GA < �-lg:XG < XG. This classification is howevernot related to the obtained stabilities, as �-lg:GA and GA stabilizedemulsions had smaller droplets than XG stabilized ones but wereless stable. A small droplet size is thus not always a guaranteeof stability and other parameters should be checked such as therheology, the probable occurrence of flocculation. . ..
Fig. 6 depicts the relationship between the retrodiffusion inten-sity of the different emulsions on the first day of stability (measuredby the Turbiscan) and the measured d43. It is obvious that theretrodiffusion intensity decreased with droplet size, as previouslysuggested (see Section 3.1.1) and in accordance with previouslyreported results [47,51].
Fig. 7. Evolution of the d43 values obtained for: (�) �-lg at 2.5 (w/w%), (�) XG at1 (w/w%), and (�) �-lg:XG (ratio 1:1) at 1 (w/w%) stabilized emulsions over a 6-month period. Continuous lines are curve fits. For each time point and each system,3 repetitions are presented.
alues obtained for these emulsions over a 6-month period of stor-ge. The graph shows no significant tendency of the d43 values toncrease over days except for XG stabilized emulsions. Indeed, d43alues of XG stabilized globules increased slightly with time. Toupport this observation, the whole set of data was analyzed with
general linear model. The covariance analysis showed that thelopes of curve fits presented on Fig. 7 were significantly differ-nt from each other. The linear model explained at 98% of the d43ariability. In a second step, a simple linear regression was testedn each formulation to determine whether there was a significantinear relationship between time and d43. For the analysis on thelope of the regression line, the significant level of 0.05 (P-value)as chosen to accept or reject the null hypothesis (i.e. the slope of
he regression line is equal to zero). Considering the XG-stabilizedmulsion, the P-value (0.008) was less than the significance level0.05). The null hypothesis was then rejected and a significant linearelationship between time and d43 can be confirmed. For emul-ions stabilized by �-lg at 2.5 (w/w%) and �-lg:XG at 1 (w/w%)-values (0.07 for �-lg and 0.89 for �-lg:XG) were higher thanhe significance level, then the null hypothesis of the slope wasot rejected. The main microscopic observations of the emulsionsFig. 5) showed no obvious droplet size variation over the 6-monthime lapse for the most stable emulsions (4 months of stabilityas considered for �-lg 2.5%). The mean diameter increase of XG
tabilized globules was not microscopically observed (as seen onig. 5(3)). However the slight increase of d43 evidenced on Fig. 7an be attributed to the enlargement of the polydispersity (resultot shown). These results demonstrate the very good stability ofhese three emulsions over the storage period; nevertheless thistability could not be explained only by the droplet size. Indeed, XGnd �-lg:XG stabilized emulsions were more stable than the �-lgne, while d43 values were in the increasing order: �-lg (2.5%) < �-g:XG (1%) < XG (1%). This order is coherent with the fact that �-lg,eing very efficient in decreasing the interfacial tension, led to theormation of smaller droplets as compared to �-lg:XG and XG sta-ilized emulsions. However, one should keep in mind that the �-lg2.5%) stabilized emulsion was differently formulated, by the use ofigh-pressure homogenizer. The use of an homogenizer evidentlyecreased �-lg stabilized droplet sizes when comparing with theon-homogenized �-lg:XG and XG stabilized ones. XG is consid-red as a non-adsorbing polysaccharide [53], which could alsoxplain why it produced larger droplets. The imposed shear dur-ng the formulation process plays an important role in the decreasef droplet sizes. d43 values obtained for �-lg:XG stabilized emul-ions were between �-lg and XG emulsions values, which seemsogical. Despite the fact that they had the smallest droplet sizes, �-g stabilized emulsions (2.5%) started to cream after about 20 days,ven if no change was observed in the corresponding d43 valuesver time.
.2. Evaluation of the stabilization mechanism
.2.1. Surface and interfacial tension measurementsSurface and interfacial tension measurements were performed
o evaluate the surface properties of the various biopolymers. Theoncentration of XG solutions was lowered to 0.1% to avoid anynfluence of bulk viscosity on the measurements. Fig. 8 depicts theime evolution of the surface tension for air-biopolymer solutions.s expected, �-lg at 2.5% decreased significantly the surface tensionf water, as the surface tension value for pure water was 72 mN/mnd the first measured value of surface tension for air- �-lg solutionas 50 mN/m. However this value changed very little over a period
f 14 h (final surface tension value was 45 mN/m). The equilibriumas rapidly reached indicating a rapid saturation of the surface. At
he initial time, the surface tension value for GA solution at 2.5% waslose to the value for pure water and the surface tension decreased
with time as GA adsorbed at the air–solution interface. The final sur-face tension value over a period of 14 h was 52 mN/m. GA neededmore time than �-lg to adsorb at the interface and decrease theinterfacial tension [27]. Unexpectedly, XG at 0.1% decreased contin-uously the surface tension from 71 mN/m to 55 mN/m. The presenceof 7 (w/w%) of proteins in the composition of the XG sample mightonly partly explain this phenomenon. Indeed, the protein concen-tration the solution of XG at 0.1 (w/w%) was very low and could notexplain the extend of this surface tension lowering. This interfacialbehaviour has rarely been described. As most of xanthan applica-tions are based on the gum ability to form high viscosity solutions,only few studies have focused on the possible surface activity of XG.However, Young et al. [54,55] observed that the surface tension ofXG solutions decreased with time. In addition, they found a rela-tionship between molecular conformation and surface properties.A combination of potentiometric titration and surface tension stud-ies showed that although the surface tension was not significantlyaffected by polymer conformation at initial times, random coils ofxanthan adsorbed faster and lowered the surface tension to a largerextent than its helix structure [54,55]. Comparing our results to thatof Young et al. [54,55] under the same condition of XG concentra-tion, the surface tension decrease was very similar when the degreeof ionization � of XG was of 0.95. One may conclude that XG hada random coil conformation in our experimental conditions. Thisis in accordance with Rochefort and Middleman [56] that reportedthat the structure of XG in distilled water at 25 ◦C is disordered orpartially ordered in the form of a randomly broken helix.
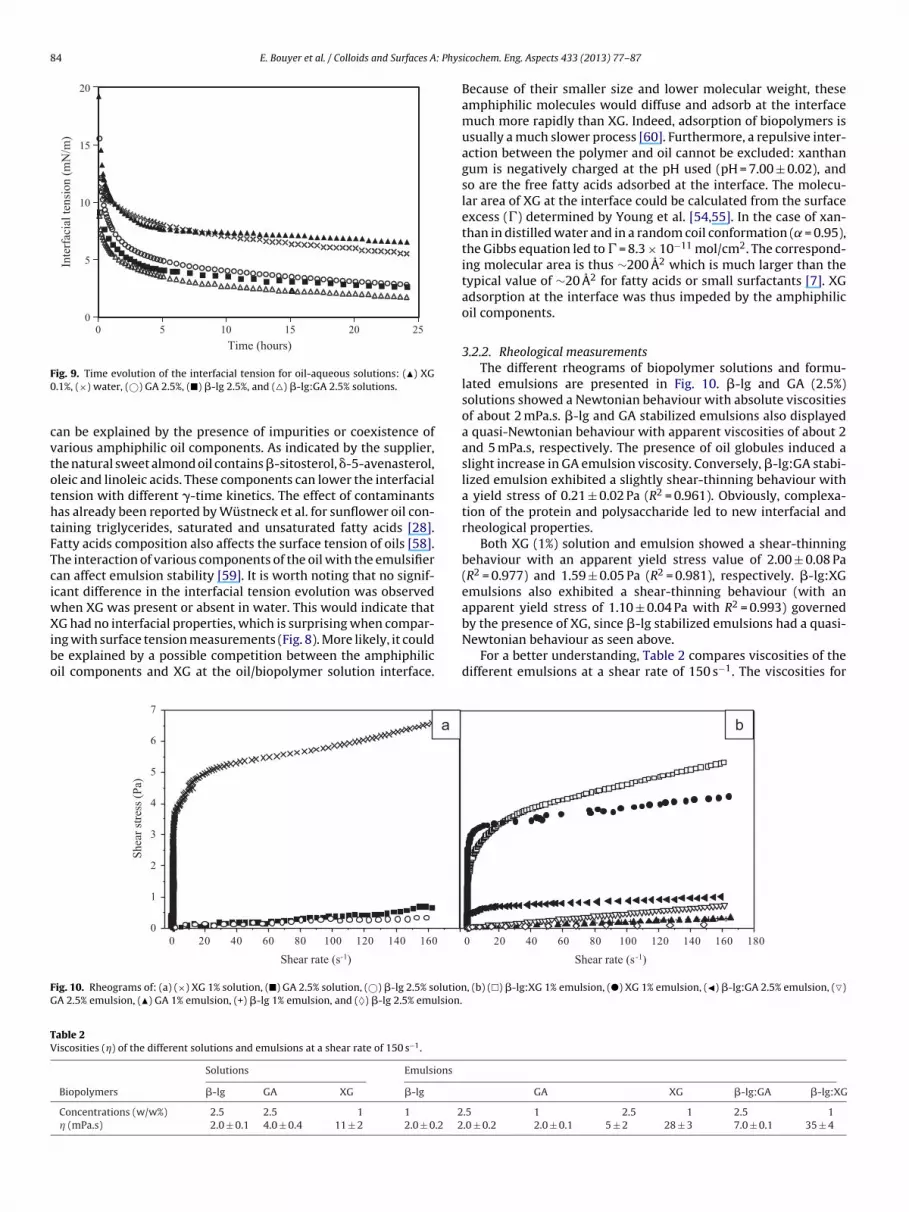
Fig. 9 represents the 24 h-interfacial tension lowering ofoil–water systems containing �-lg and GA at 2.5%, �-lg:GA at 2.5%,XG at 0.1%, or only pure water. �-lg, GA, and �-lg:GA solutions sig-nificantly decreased the interfacial tension. �-lg adsorbed fasterthan GA. At the initial time (t = 0 h), the interfacial tension valuesfor �-lg and GA solutions were about 9 and 16 mN/m, respectively.However, after few hours, their interfacial activity appeared simi-lar. �-lg:GA mixed solutions showed the best interfacial properties,probably due to the formation of the �-lg:GA complex. It has beenpreviously shown that protein:polysaccharide electrostatic com-plexes exhibited improved interfacial properties compared to thesingle components [33,57]. However, as observed before, �-lg:GAstabilized emulsions phase separated within 3 days. Excess free andcomplexed biopolymers in the aqueous phase probably influenced
the stabilization mechanisms by inducing depletion or bridgingflocculation [27].
It is interesting to note that the pure oil–water interfacial ten-sion decreased from 21 to 6 mN/m over a 24 h-period. This decrease
an be explained by the presence of impurities or coexistence ofarious amphiphilic oil components. As indicated by the supplier,he natural sweet almond oil contains �-sitosterol, �-5-avenasterol,leic and linoleic acids. These components can lower the interfacialension with different �-time kinetics. The effect of contaminantsas already been reported by Wüstneck et al. for sunflower oil con-aining triglycerides, saturated and unsaturated fatty acids [28].atty acids composition also affects the surface tension of oils [58].he interaction of various components of the oil with the emulsifieran affect emulsion stability [59]. It is worth noting that no signif-cant difference in the interfacial tension evolution was observed
hen XG was present or absent in water. This would indicate thatG had no interfacial properties, which is surprising when compar-
ng with surface tension measurements (Fig. 8). More likely, it coulde explained by a possible competition between the amphiphilicil components and XG at the oil/biopolymer solution interface.
Because of their smaller size and lower molecular weight, theseamphiphilic molecules would diffuse and adsorb at the interfacemuch more rapidly than XG. Indeed, adsorption of biopolymers isusually a much slower process [60]. Furthermore, a repulsive inter-action between the polymer and oil cannot be excluded: xanthangum is negatively charged at the pH used (pH = 7.00 ± 0.02), andso are the free fatty acids adsorbed at the interface. The molecu-lar area of XG at the interface could be calculated from the surfaceexcess (�) determined by Young et al. [54,55]. In the case of xan-than in distilled water and in a random coil conformation ( = 0.95),the Gibbs equation led to � = 8.3 × 10−11 mol/cm2. The correspond-ing molecular area is thus ∼200 A2 which is much larger than thetypical value of ∼20 A2 for fatty acids or small surfactants [7]. XGadsorption at the interface was thus impeded by the amphiphilicoil components.
3.2.2. Rheological measurementsThe different rheograms of biopolymer solutions and formu-
lated emulsions are presented in Fig. 10. �-lg and GA (2.5%)solutions showed a Newtonian behaviour with absolute viscositiesof about 2 mPa.s. �-lg and GA stabilized emulsions also displayeda quasi-Newtonian behaviour with apparent viscosities of about 2and 5 mPa.s, respectively. The presence of oil globules induced aslight increase in GA emulsion viscosity. Conversely, �-lg:GA stabi-lized emulsion exhibited a slightly shear-thinning behaviour witha yield stress of 0.21 ± 0.02 Pa (R2 = 0.961). Obviously, complexa-tion of the protein and polysaccharide led to new interfacial andrheological properties.
Both XG (1%) solution and emulsion showed a shear-thinningbehaviour with an apparent yield stress value of 2.00 ± 0.08 Pa(R2 = 0.977) and 1.59 ± 0.05 Pa (R2 = 0.981), respectively. �-lg:XGemulsions also exhibited a shear-thinning behaviour (with anapparent yield stress of 1.10 ± 0.04 Pa with R2 = 0.993) governed
Newtonian behaviour as seen above.For a better understanding, Table 2 compares viscosities of the
different emulsions at a shear rate of 150 s−1. The viscosities for
E. Bouyer et al. / Colloids and Surfaces A: Physicochem. Eng. Aspects 433 (2013) 77– 87 85
F h (a) �a
tledtctaTsc
sbi
�
wititay(
mweo1swvrsc–rcr
3
css(efgc
ig. 11. Schematic depiction of the different obtained stabilization mechanisms witt pH 7.
he various emulsions can be classified in the order: �-lg < GA < �-g:GA < XG < �-lg:XG. These values explain the better stability ofmulsions containing XG compared to �-lg alone, even if theroplets formed in the �-lg stabilized emulsions were smaller thanhose in XG stabilized ones. XG clearly enhanced the viscosity of theontinuous phase of the emulsions and conferred them a shear-hinning behaviour. Thus, the apparent viscosity was rather hight low shear stress and decreased when the stress was increased.his behaviour has largely been reported in the literature for XGtabilized emulsions where it is used to prevent flocculation andreaming [14,44,61–63].
Some authors have estimated the minimal value of the yieldtress (�0) of the weakly viscosified aqueous phase, required to sta-ilize droplets and prevent creaming in the emulsions, as reported
n Eq. (2) [14,63].
0 ≥ 43
�gR (2)
ith � the density difference between the two phases, g the grav-ty acceleration, and R the mean radius of the droplets. Knowinghe mean radius allows the determination of the necessary min-mum yield stress value to obtain for stable emulsions. Applyinghis equation for XG, �-lg:XG, and �-lg:GA stabilized emulsions canllow us to predict the stability of these emulsions. If the apparentield stress obtained experimentally is above the theoretical onecalculated from Eq. (2)), emulsions can be considered as stable.
If we assume a density () of about 1 × 103 kg/m3 for all biopoly-er solutions and of 0.9 × 103 kg/m3 for the oil phase used in thisork, � ≈ 100 kg/m3. The gravity acceleration is considered as
qual to 10 m/s2 and the mean radius is calculated on the basisf the obtained d43 values. The following values were thus used:8.5, 9, and 3.5 �m for XG, �-lg:XG, and �-lg:GA stabilized emul-ions, respectively. The theoretical minimum yield stress valuesere 0.025, 0.012, and 0.005 Pa, respectively. If one compares these
alues with those obtained experimentally: 1.59, 1.10, and 0.21 Pa,espectively, all the emulsions should be stable. However, �-lg:GAtabilized emulsion phase separated very quickly. An explanationould be that the condition needed to apply the previous equation
i.e. globules are considered as isolated spheres – is probably notespected. This is further evidence that some flocculation and/oroalescence probably occurred, in accordance with our previousesults (microscopy).
.2.3. General discussionTaking into account all the results obtained in this study and
ombining them with results obtained previously [27], differenttabilization mechanisms have been identified for biopolymer-tabilized emulsions (Fig. 11). In �-lg stabilized emulsionsFig. 11(a)), the mechanism by which the protein stabilized the
mulsions is well known. �-lg adsorbed at the oil–water inter-ace, rearranged its structure in order to expose hydrophobicroups to the lipophilic phase [25,26], and decreased the interfa-ial tension [27,64]. Moreover, because the protein was positively
-lg at pH 4.2, (b) GA at pH 4.2, (c) XG at pH 7, (d) �-lg:GA at pH 4.2, and (e) �-lg:XG
charged at pH 4.2, electrostatic repulsions occurred between oildroplets thus contributing to the stabilization. It has also beenreported that adsorbed �-lg films possess an elastic behaviour[27,28]. Wüstneck et al., performed interfacial dilational rheologymeasurements using the pendant drop method and showed thatlow protein concentrations led to an optimally packed film of �-lgmolecules [28]. On the contrary, at high concentrations, abun-dant lateral protein–protein interactions induced a more compactconformation of the protein at the interface. We made similarobservations in our previous work [27], showing that 1 and 2.5%concentrations were high compared to the protein concentrationnecessary for interfacial saturation (7 × 10−4 (w/w%)) [27]. In theemulsions studied in the present work, �-lg molecules in excesswere free in the bulk aqueous phase. These emulsions startedcreaming after about 20 days as evidenced by Turbiscan measure-ments, probably because of non-optimal interfacial packing or bya depletion flocculation mechanism.
For the stabilization mechanism of emulsions by GA (Fig. 11(b)),interfacial measurements confirmed that the gum was able toadsorb at the oil–water interface and decrease the interfacial ten-sion. Interfacial adsorption most probably occurred via the AGPfraction of GA: the proteinaceous component would adsorb at theoil/water interface and the carbohydrate component would extendout from droplets surface into the aqueous phase [35,39].
Despite its obvious adsorption at the air/solution interface, XGproved unable to lower the sweet almond oil/XG solution interface.It appeared thus as a poor surfactant at the oil/water interface. Withregard to rheological measurements, it seems that oil-dropletsstability can primarily be attributed to the high viscosity and shear-thinning behaviour of the aqueous phase, i.e. to the network formedby XG molecules (Fig. 11(c)). The high viscosity at low shear rateprevents droplets from encountering each other and thus slowsdown flocculation and creaming mechanisms.
The next identified stabilization mechanism is that for �-lg:GAstabilized emulsions (Fig. 11(d)). Taking into account the resultsobtained in [27], it may be concluded that thanks to attractiveelectrostatic interactions, GA can form a second layer around oildroplets already covered by �-lg. This bi-layer stabilization mech-anism is often reported to enhance emulsion stability compared to abiopolymer alone [19,20]. However, in our case, �-lg:GA stabilizedemulsions presented a creaming within 3 days. Further experi-ments are thus required to optimize the biopolymer concentrationsto avoid flocculation.
The last mechanism is the one observed for �-lg:XG stabilizedemulsions (Fig. 11(e)). If �-lg molecules first adsorbed at the inter-face, the addition of XG enhanced emulsion stability from 20 daysto more than 6 months by modifying the continuous phase rhe-ology, without reducing the droplet size. At pH = 7, �-lg was wellabove its pI and was highly negatively charged. As XG was also
negatively charged, no complexation could occur between the twobiopolymers. Perez et al. [65] showed that at pH 7, �-lg adsorp-tion in the presence of XG could be enhanced by a mechanismbased on biopolymer segregative interactions mainly due to the
lectrostatic repulsions between the two biopolymers and thermo-ynamic incompatibility in the interface vicinity, resulting in betterurface and viscoelastic properties. It is a very interesting result ast shows the possibility to obtain biopolymer stabilized emulsions
ithout adding any synthetic surfactant, nor using a high pressureomogenizer.
. Conclusions
We describe a stability study for emulsions stabilized with �-g, GA, XG, �-lg:GA, and �-lg:XG. Emulsions stabilized by GA and-lg:GA exhibited the lowest stability. �-lg stabilized emulsionshase separated after about 20 days. Conversely, emulsions con-aining XG did not show any phase separation for the studied-month period. The stabilization mechanisms were deduced from
nterfacial tension and rheology measurements, and microscopicbservations. XG showed limited surface properties, especially atil/solution interface, but it formed a polymer network in waterith a high yield stress and it contributed to the shear-thinning
ehaviour of the emulsions. Its capacity to entrap the emul-ion droplets would explain the longer stability of its emulsions,ompared to �-lg and GA stabilized emulsions, which exhibiteduasi-Newtonian behaviour with a lower viscosity.
Different stabilization mechanisms were identified. Whereasoth �-lg and GA adsorbed at the interface, the lengthier adsorp-ion of GA impairs emulsion stability. When mixing �-lg and GA, aouble interfacial layer was formed by interfacial adsorption of therotein, followed by the polysaccharide binding through attractivelectrostatic interactions. XG stabilized oil droplets mainly throughhe modification of emulsion rheological properties. Finally, the last
echanism is that of a �-lg adsorbed layer at the oil–water interfacend free XG molecules in the aqueous phase modifying emulsionheology. This work demonstrates that by varying the nature of theiopolymers and their combination, it is possible to obtain various
nterfacial and rheological behaviours. Those properties can be usedo formulate new pharmaceutical emulsions without synthetic sur-actant and more respectful towards sustainable development.
cknowledgments
We thank Morgane Beigneux for her technical assistance in thexperimental part of the work and Dr Anouar Benmalek for theruitful discussions regarding the statistical analysis of the data.
eferences
[1] N. Garti, What can nature offer from an emulsifier point of view: trends andprogress, Colloids Surf., A 152 (1999) 125–146.
[2] R. Lutz, A. Aserin, L. Wicker, N. Garti, Double emulsions stabilized by a chargedcomplex of modified pectin and whey protein isolate, Colloids Surf. B 72 (2009)121–127.
[3] B.F. McNamee, E.D. O’Riordan, M. O’Sullivan, Emulsification and microencap-sulation properties of gum arabic, J. Agric. Food Chem. 46 (1998) 4551–4555.
[6] X. Huang, Y. Kakuda, W. Cui, Hydrocolloids in emulsions: particle size distribu-tion and interfacial activity, Food Hydrocolloids 15 (2001) 533–542.
[7] E. Bouyer, G. Mekhloufi, V. Rosilio, J.L. Grossiord, F. Agnely, Proteins, polysac-charides, and their complexes used as stabilizers for emulsions: Alternativesto synthetic surfactants in the pharmaceutical field? Int. J. Pharm. 436 (1–2)(2012) 359–378.
[8] T. Cserháti, E. Forgács, G. Oros, Biological activity and environmental impact ofanionic surfactants, Environment International 28 (2002) 337–348.
[9] I. Effendy, H.I. Maibach, Surfactants and experimental irritant contact dermati-tis, Contact Dermatitis 33 (1995) 217–225.
10] E. Liwarska-Bizukojc, K. Miksch, A. Malachowska-Jutsz, J. Kalka, Acute toxicityand genotoxicity of five selected anionic and non-ionic surfactants, Chemo-sphere 58 (2005) 1249–1253.
[
[
icochem. Eng. Aspects 433 (2013) 77– 87
11] E. Dickinson, Proteins at liquid interfaces, in: K.L. Mittal, Promod Kumar (Eds.),An Introduction to Food Colloids, Oxford University Press, New York, 1992, pp.140–173.
12] E. Dickinson, Hydrocolloids as emulsifiers and emulsion stabilizers, FoodHydrocolloids 23 (2009) 1473–1482.
13] J. Weiss, I. Scherze, G. Muschiolok, Polysaccharide gel with multiple emulsion,Food Hydrocolloids 19 (2005) 605–615.
14] M. Benna-Zayani, N. Kbir-Ariguib, M. Trabelsi-Ayadi, J.-L. Grossiord, Stabiliza-tion of W/O/W double emulsion by polysaccharides as weak gels, Colloids Surf.,A 316 (2008) 46–54.
15] I. Capek, Degradation of kinetically-stable o/w emulsions, Adv. Colloid InterfaceSci. 107 (2004) 125–155.
16] E. Dickinson, Emulsion stabilization by polysaccharides and protein–polysaccharide complexes, in: A.M. Stephen (Ed.), Food Polysaccharides andTheir Applications, Marcel Dekker, New York, 1995, pp. 501–515.
17] E. Dickinson, Emulsification and emulsion stabilization with protein–polysaccharide complexes, in: P.A. Williams, G.O. Phillips (Eds.), Gums andStabilizers for the Food Industry-14, Royal Society, Cambridge, UK, 2008, pp.221–232.
18] E. Dickinson, Interfacial structure and stability of food emulsions as affected byprotein-polysaccharide interactions, Soft Matter 4 (2008) 932–942.
19] D. Guzey, D.J. McClements, Formation, stability and properties of multilayeremulsions for application in the food industry, Adv. Colloid Interface Sci. 128(2006) 227–248.
20] Y.S. Gu, E.A. Decker, D.J. McClements, Application of multi-component biopoly-mer layers to improve the freeze–thaw stability of oil-in-water emulsions:�-lactoglobulin-�-carrageenan-gelatin, J.Food Eng. 80 (2007) 1246–1254.
21] P. Thanasukarn, R. Pongsawatmanit, D.J. McClements, Utilization of layer-by-layer interfacial deposition technique to improve freeze-thaw stability of oil-in-water emulsions, Food Res. Int. 39 (6) (2006) 721–729.
22] E. Bouyer, Stabilisation d’émulsions d’intérêt pharmaceutique par des protéineset des polysaccharides: exemples de la �-lactoglobuline, de la gomme arabiqueet de la gomme xanthane, Université Paris Sud, France, 2011 (PhD thesis).
24] E. Dickinson, Properties of emulsions stabilized with milk proteins: overviewof some recent developments, J.Dairy Sci. 80 (1997) 2607–2619.
25] S. Damodaran, Protein stabilization of emulsions and foams, J.Food Sci. 70(2005) 54–66.
26] M.A. Bos, T.V. Vliet, Interfacial rheological properties of adsorbed protein layersand surfactants: a review, Adv. Colloid Interface Sci. 91 (2001) 437–471.
27] E. Bouyer, G. Mekhloufi, I. Le Potier, T. du Fou de Kerdaniel, J.-L. Grossiord,V. Rosilio, F. Agnely, Stabilization mechanism of oil-in-water emulsionsby �-lactoglobulin and gum arabic, J. Colloid Interface Sci. 354 (2011)467–477.
28] R. Wüstneck, B. Moser, G. Muschiolik, Interfacial dilational behaviour ofadsorbed �-lg layers at different fluid interfaces, Colloids Surf. B 15 (1999)263–273.
29] E. Dickinson, Proteins at interfaces and in emulsions stability, rheology andinteractions, J. Chem. Soc. Faraday Trans. 94 (12) (1998) 1657–1669.
30] P. Wilde, A. Mackie, F. Husband, P. Gunning, V. Morris, Proteins and emulsifiersat liquid interfaces, Adv. Colloid Interface Sci. 108-109 (2004) 63–71.
31] D.M.W. Anderson, M.M.E. Bridgeman, J.G.K. Farquhar, C.G.A. McNab, The chemi-cal characterization of the test article used in toxicological studies of gum arabic(Acacia Senegal (L.) Willd), Int. Tree Crops 2 (1983) 245–254.
32] O.H.M. Idris, P.A. Williams, G.O. Phillips, Characterization of gum from Acaciasenegal trees of different age and location using multidetection gel permeationchromatography, Food Hydrocolloids 12 (1998) 379–388.
33] R.C. Randall, G.O. Phillips, P.A. Williams, The role of the proteinaceous compo-nent on the emulsifying properties of gum arabic, Food Hydrocolloids 2 (1988)131–140.
34] P. Erni, E.J. Windhab, R. Gunde, M. Graber, B. Pfister, A. Parker, P. Fischer,Interfacial rheology of surface-active biopolymers: Acacia senegal gumversus hydrophobically modified starch, Biomacromolecules 8 (11) (2007)3458–3466.
35] L. Picton, I. Bataille, G. Muller, Analysis of a complex polysaccharide (gum ara-bic) by multi-angle laser light scattering coupled on-line to size exclusionchromatography and flow field flow fractionation, Carbohydr. Polym. 42 (1)(2000) 23–31.
36] M.L. Fauconnier, C. Blecker, J. Groyne, H. Razafindralambo, E. Vanzeveren, M.Marlier, M. Paquot, Characterization of two Acacia gums and their fractionsusing a Langmuir film balance, J. Agric. Food Chem. 48 (7) (2000) 2709–2712.
37] E. Dickinson, D.J. Elverson, B.S. Murray, On the film-forming and emulsion-stabilizing properties of gum Arabic: dilution and flocculation aspects, FoodHydrocolloids 3 (1989) 101–114.
38] E. Dickinson, Hydrocolloids at interfaces and the influence on the properties ofdispersed systems, Food Hydrocolloids 17 (2003) 25–39.
39] R. Chanamai, D.J. McClements, Comparison of gum arabic, modified starch, andwhey protein isolate as emulsifiers: influence of pH, CaCl2 and temperature, J.Food Sci. 67 (1) (2002) 120–125.
40] J.A. Casas, V.E. Santos, F. Garcia-Ochoa, Xanthan gum production under sev-eral operational conditions: molecular structure and rheological properties,Enzyme Microb. Technol. 26 (2000) 282–291.
41] M. Milas, M. Rinaudo, Conformational investigation on the bacterial polysac-charide xanthan, Carbohydr. Res. 76 (1979) 189–196.
42] P.-E. Jansson, L. Kenne, B. Lindberg, Structure of the extracellular polysaccharidefrom Xanthomonas campestris, Carbohydr. Res. 45 (1975) 275–282.
43] L.D. Melton, L. Mindt, D.A. Rees, G.R. Sanderson, Covalent structure of the extra-cellular polysaccharide from Xanthomonas campestris: evidence from partialhydrolysis studies, Carbohydr. Res. 46 (1976) 245–257.
44] C. Sun, S. Gunasekaran, M.P. Richards, Effect of xanthan gum on physicochem-ical properties of whey protein isolate stabilized oil-in-water emulsions, FoodHydrocolloids 21 (2007) 555–564.
45] G. Mekhloufi, C. Sanchez, D. Renard, S. Guillemin, L. Hardy, pH-Induced struc-tural transitions during complexation and coacervation of beta-lactoglobulinand acacia gum, Langmuir 21 (2005) 386–394.
46] C. Schmitt, Etude de la coacervation complexe entre la �-lactoglobuline et lagomme d’acacia en solution aqueuse, Institut National Polytechnique de Lor-raine, Nancy, France, 2000 (Ph.D thesis).
47] O. Mengual, G. Meunier, I. Cayré, K. Puech, P. Snabre, TURBISCAN MA 2000: mul-tiple light scattering measurements for concentrated emulsion and suspensioninstability analysis, Talanta 50 (1999) 445–456.
48] C. Lemarchand, P. Couvreur, C. Vauthier, D. Costantini, R. Gref, Study of emulsionstabilization by graft copolymers using the optical analyzer Turbiscan, Int. J.Pharm. 254 (2003) 77–82.
49] C. Ringard-Lefebvre, A. Bochot, E. Memisoglu, D. Charon, D. Duchêne, A. Baszkin,Effect of spread amphiphilic �-cyclodextrins on interfacial properties of theoil/water system, Colloids Surf. B 25 (2002) 109–117.
50] M. El-Mahrab-Robert, V. Rosilio, M.A. Bolzinger, P. Chaminade, J.-L. Grossiord,Assessment of oil polarity: comparison of evaluation methods, Int. J. Pharm.348 (2008) 89–94.
51] H. Buron, O. Mengual, G. Meunier, I. Cayré, P. Snabre, Optical characterizationof concentrated dispersions: applications to laboratory analyses and on-lineprocess monitoring and control, Polym. Int. 53 (2004) 1205–1209.
52] A. Pizzino, M. Catté, E. Van Hecke, J-L. Salager, J-M. Aubry, On-line light
backscattering tracking of the transitional phase inversion of emulsions, Col-loids Surf., A 338 (2009) 148–154.
53] T. Moschakis, B.S. Murray, E. Dickinson, Particle tracking using confocalmicroscopy to probe the microrheology in a phase-separating emulsion con-taining nonadsorbing polysaccharide, Langmuir 22 (2006) 4710–4719.
[
icochem. Eng. Aspects 433 (2013) 77– 87 87
54] S.-L. Young, Effect of molecular conformation on the surface properties of poly-electrolyte solutions: Xanthan gum model studies, Oregon State University,Corvallis, OR, 1987 (ScM thesis).
55] S-L. Young, J.A. Torres, Xanthan: effect of molecular conformation on surfacetension properties, Food Hydrocolloids 3 (5) (1989) 365–377.
56] W.E. Rochefort, S. Middleman, Rheology of xanthan gum: salt, temperature, andstrain effects in oscillatory and steady shear experiments, J. Rheol. 31 (1987)337–369.
57] V. Ducel, J. Richard, Y. Popineau, F. Boury, Rheological interfacial propertiesof plant protein–arabic gum coacervates at the oil–water interface, Biomacro-molecules 6 (2005) 790–796.
58] C.A.W. Allen, K.C. Watts, R.G. Ackman, Predicting the surface tension ofbiodiesel fuels from their fatty acid composition, JAOCS 76 (3) (1999)317–323.
59] F. Cournarie, M.-P. Savelli, V. Rosilio, F. Bretez, C. Vauthier, J.-L. Grossiord, M.Seiller, Insulin-loaded W/O/W multiple emulsions: comparison of the perform-ances of systems prepared with medium-chain-triglycerides and fish oil, Eur.J. Pharm. Biopharm. 58 (2004) 477–482.
60] E. Dickinson, Mixed biopolymers at interfaces: competitive adsorption andmultilayer structures, Food Hydrocolloids 25 (2011) 1966–1983.
61] M. Hennock, R.R. Rahalkar, P. Richmond, Effect of xanthan gum upon therheology and stability of oil–water emulsions, J. Food Sci. 49 (5) (1984)1271–1274.
62] R.P. Chhabra, Bubbles Drops and Particles in Non-Newtonian Fluids, CRC Press,Boca Raton, FL, 1993.
63] L. Jossic, A. Magnin, Structuring of gelled suspensions flowing through a sud-den three dimensional expansion, J. Non-Newtonian Fluid Mech. 127 (2005)201–212.
64] M. Paulsson, P. Dejmek, Surface film pressure of �-lactoglobulin, �-lactalbuminand bovine serum albumin at the air/water interface studied by Wilhelmy plate
and drop volume, J. Colloid Interface Sci. 150 (2) (1992) 394–403.
65] A.A. Perez, C. Carrera Sanchez, J.M. Rodriguez Patino, A.C. Rubiolo, L.G. Santiago,Milk whey proteins and xanthan gum interactions in solution and at the air-water interface: a rheokinetic study, Colloids and Surfaces B: Biointerfaces 81(2010) 50–57.