From uncoupled to coupled Hartree-Fock polarizabilities of infinite polymeric chains. Pariser-Parr-Pople applications to the polyacetylene chains BenoTt Champagne, a) Joseph G. Fripiat, and Jean-Marie Andre Laboratoire de Chimie Th&ori$ue Appliqute, FacuMs Universitaires Notre-Dame de la Paix, 61. rue de Bruxelles, B-So00 Namur, Belgium (Received22 November 1991;accepted18 February 1992) A generalmethod is formulated to compute the asymptotic longitudinal polarizabilities of infinite systems. This procedureis an extensionto infinite systems of the molecular random- phase-approximation method which provides coupled Hartree-Fock valuesand thus takes into account the field-induced electron reorganizationaleffects.It is shown that the Genkin- Mednis uncoupled method corresponds to the drastic approximation of the coupled one where there is no electron reorganization. By looking at the asymptotic polarizabilities as the convergence values of the oligomeric results, the method is tested for polyacetylene chains in the Pariser-Parr-Pople approximation. 1. INTRODUCTION Materials which exhibit high linear and nonlinear re- sponses are currently the subject of intense investigations from a decade.’Large organic conjugated molecules are among the most interesting ones. Indeed, these molecules present high electric susceptibilities which depend very much on the nature of the delocalized network of s- or u electrons.*The advantage of organic systems lies in the fact that their molecular structure can be easily modified in order to maximize their electric response. Quantum chemistry and its methodsmay predict the linear and nonlinear responses and thus may orientate the synthesisand accelerate the re- search. Since polyacetylene (PA) can be considered as the pro- totype of extended?rsystems”‘* and since the evolution of the optical properties of PA chains with respectto their size hasbeen extensivelystudied, we present for the sakeof com- pleteness a summary of existing calculations. They all dem- onstrate the need of getting accurateasymptotic values. For example, Hameka3 obtained the analytical expres- sions for the Hiickel static longitudinal polarizabilities of finite and regular polyacetylenechains by using the pertur- bation theory. In a recent paper,‘* we have extended Hame- ka’s procedureto the altemant chain and studied the influ- ence of the bond alternation on the polarizability of these systems. In the samestudy, the Genkin-Mednis approachI was used to calculate the frequency-independent longitudi- nal polarizability per unit cell for the corresponding (alter- nant or regular) infinite systems.This Genkin-Mednis ap- proach has already been used by Cojan, Agrawal, and Flytzanis14to describethe optical properties of one-dimen- sional ( 1D) semiconductors and of conjugated polymers and to calculate the polarizabilities of polyacetylene and po- lydiacetylene chains. They also introduced the concept of delocalizationlength (extent of the electron delocalization) . “Research Assistant of the National Fund for Scientific Research, Bel- gium. However, the Hiickel methodology presentsan impor- tant drawback since it doesnot take electron-electron Cou- lomb interactions explicitly into account. On the other hand, the iterative Hartree-Fock method at different levels of ap- proximation takes care of theseCoulomb interactions. Ha- meka and Zamani-Khamiri4 used a valence perturbation methodi5,i6 (VPT) coupled with the Pariser-Parr- Pople’7-‘9 (PPP) approximation in order to compare these VPT-PPP polarizabilities with the values obtained in the Hiickel calculations. The inclusion of the Coulomb terms inducesa decrease of 1 order of magnitude for the longitudi- nal polarizabilities. Soos and Hayden” have reached the same conclusion by using a PPP-valence-bond technique. By extrapolation of the curve representing the inverseof the longitudinal polarizability per site (N/a, ) with respectto the inverse of the number of sites ( l/N), they have found that the asymptotic values of a,/N for N equal to infinity are, respectively, 50 a.u. per carbon atom at the PPP level and 641 a.u. at the Hiickel level. de Melo and Silbey’ have calculated the longitudinal polarizability of polyacetylene chains by using the perturbative expansion of the density- matrix (PEDM) method of McWeeny*’ in the framework of the PPP approximation. For oligomers ranging from 2 to 20 carbon atoms, they derived an expression for the longitu- dinal polarizability per carbon atom with respect to the number of carbon atoms: a,/N = 4.63N0.” a.u. The same method was usedby Papadopoulos, Waite, and Nicolaides’ in the framework of CND0/2 approximations** to calcu- late the average polarizability of the four first polyene oli- gomers (ethylene, butadiene, hexatriene,and octatetraene). In this work, where the basis-set parameters were optimized to fit the static average experimental polarizability of ethyl- ene, the results are just three times greater than those of de Melo and Silbey. More recently, K&man’ determined static longitudinal polarizabilities ((r, ) at the intermediate ne- glect of differential overlap (INDO) level** of approxima- tion for finite polyene chain models of polyacetylene con- taining up to 62 carbon atoms (N = 3 1) . By extrapolation, 8330 J. Chem. Phys. 96 (1 l), 1 June 1992 0021-9606/92/l 18330-08$06.00 @ 1992 American Institute of Physics
Transcript
From uncoupled to coupled Hartree-Fock polarizabilities of infinite polymeric chains. Pariser-Parr-Pople applications to the polyacetylene chains
BenoTt Champagne, a) Joseph G. Fripiat, and Jean-Marie Andre Laboratoire de Chimie Th&ori$ue Appliqute, FacuMs Universitaires Notre-Dame de la Paix, 61. rue de Bruxelles, B-So00 Namur, Belgium
(Received 22 November 1991; accepted 18 February 1992)
A general method is formulated to compute the asymptotic longitudinal polarizabilities of infinite systems. This procedure is an extension to infinite systems of the molecular random- phase-approximation method which provides coupled Hartree-Fock values and thus takes into account the field-induced electron reorganizational effects. It is shown that the Genkin- Mednis uncoupled method corresponds to the drastic approximation of the coupled one where there is no electron reorganization. By looking at the asymptotic polarizabilities as the convergence values of the oligomeric results, the method is tested for polyacetylene chains in the Pariser-Parr-Pople approximation.
1. INTRODUCTION
Materials which exhibit high linear and nonlinear re- sponses are currently the subject of intense investigations from a decade.’ Large organic conjugated molecules are among the most interesting ones. Indeed, these molecules present high electric susceptibilities which depend very much on the nature of the delocalized network of s- or u electrons.* The advantage of organic systems lies in the fact that their molecular structure can be easily modified in order to maximize their electric response. Quantum chemistry and its methods may predict the linear and nonlinear responses and thus may orientate the synthesis and accelerate the re- search.
Since polyacetylene (PA) can be considered as the pro- totype of extended ?r systems”‘* and since the evolution of the optical properties of PA chains with respect to their size has been extensively studied, we present for the sake of com- pleteness a summary of existing calculations. They all dem- onstrate the need of getting accurate asymptotic values.
For example, Hameka3 obtained the analytical expres- sions for the Hiickel static longitudinal polarizabilities of finite and regular polyacetylene chains by using the pertur- bation theory. In a recent paper,‘* we have extended Hame- ka’s procedure to the altemant chain and studied the influ- ence of the bond alternation on the polarizability of these systems. In the same study, the Genkin-Mednis approachI was used to calculate the frequency-independent longitudi- nal polarizability per unit cell for the corresponding (alter- nant or regular) infinite systems. This Genkin-Mednis ap- proach has already been used by Cojan, Agrawal, and Flytzanis14 to describe the optical properties of one-dimen- sional ( 1D) semiconductors and of conjugated polymers and to calculate the polarizabilities of polyacetylene and po- lydiacetylene chains. They also introduced the concept of delocalization length (extent of the electron delocalization) .
“Research Assistant of the National Fund for Scientific Research, Bel- gium.
However, the Hiickel methodology presents an impor- tant drawback since it does not take electron-electron Cou- lomb interactions explicitly into account. On the other hand, the iterative Hartree-Fock method at different levels of ap- proximation takes care of these Coulomb interactions. Ha- meka and Zamani-Khamiri4 used a valence perturbation methodi5,i6 (VPT) coupled with the Pariser-Parr- Pople’7-‘9 (PPP) approximation in order to compare these VPT-PPP polarizabilities with the values obtained in the Hiickel calculations. The inclusion of the Coulomb terms induces a decrease of 1 order of magnitude for the longitudi- nal polarizabilities. Soos and Hayden” have reached the same conclusion by using a PPP-valence-bond technique. By extrapolation of the curve representing the inverse of the longitudinal polarizability per site (N/a, ) with respect to the inverse of the number of sites ( l/N), they have found that the asymptotic values of a,/N for N equal to infinity are, respectively, 50 a.u. per carbon atom at the PPP level and 641 a.u. at the Hiickel level. de Melo and Silbey’ have calculated the longitudinal polarizability of polyacetylene chains by using the perturbative expansion of the density- matrix (PEDM) method of McWeeny*’ in the framework of the PPP approximation. For oligomers ranging from 2 to 20 carbon atoms, they derived an expression for the longitu- dinal polarizability per carbon atom with respect to the number of carbon atoms: a,/N = 4.63N0.” a.u. The same method was used by Papadopoulos, Waite, and Nicolaides’ in the framework of CND0/2 approximations** to calcu- late the average polarizability of the four first polyene oli- gomers (ethylene, butadiene, hexatriene, and octatetraene). In this work, where the basis-set parameters were optimized to fit the static average experimental polarizability of ethyl- ene, the results are just three times greater than those of de Melo and Silbey. More recently, K&man’ determined static longitudinal polarizabilities ((r, ) at the intermediate ne- glect of differential overlap (INDO) level** of approxima- tion for finite polyene chain models of polyacetylene con- taining up to 62 carbon atoms (N = 3 1) . By extrapolation,
8330 J. Chem. Phys. 96 (1 l), 1 June 1992 0021-9606/92/l 18330-08$06.00 @ 1992 American Institute of Physics
he obtained an estimated value of 110.5 a.u. for a,/N as N tends to infinity. These semiempirical calculations demon- strate the large sensitivity of the computed values of polari- zabilities and hyperpolarizabilities to the parametrization used in the semiempirical methods.
At the ab initio level and using the finite-field (FF) tech- nique,23 Bodart et aL6 have analyzed the behavior of the longitudinal polarizability of polyacetylene chains (from ethylene to all-trans-octatetraene) with respect to the extent of the basis set [minimal STO-3G (Ref. 24) or split valence 4-3 1G (Ref. 25) 1. The scaling factors between these two basis sets and the experimental values were set in such a way that they are able to obtain calculated values close to experi- mental ones. They have extended this study by looking at the influence of the bond and chain length on the longitudinal polarizability of polyenes ranging from 2 to 20 carbon atoms.* Using the ab initio coupled perturbed Hat-tree-Fock (CPHF) technique, 26 Hurst, Dupuis, and Clementi’ have computed the static dipole longitudinal (a, ) and averaged (a) polarizabilities of the polyenes ranging from C,H, to
c22 H24 * Minimal STO-3G and extended 6-31G,” 6- 31G*,25 and 6-31G + PD basis-set calculations show the behavior of a/N with respect to N (N being the number of carbon pairs). These authors have fitted the obtained values by an equation of the type log (a/N) = a + b /N + c/N ‘. In such an equation, the saturation pattern of the polarizability per unit cell is included in contrast with the expression of de Melo and Silbey, which only characterized the nonlinear pattern of a/N for the first oligomers. The limiting value of the polarizability per unit cell with N going to infinity is obviously given by the following expression:
lim ; = 10”. (1) N-CO
Using this relation, Hurst, Dupuis, and Clementi obtained values (expressed in a.u.) of 38.9,60.3,57.0, and 60.8 for the average polarizability and 105.9, 163.3, 154.8, and 160.5 for the longitudinal polarizability for the STO-3G, 6-31G, 6- 3 lG*, and 6-3 IG + PD basis sets, respectively. Recently, Kurtz” compared the results obtained by the FF method using semiempirical modified neglect of differential overlap (MNDO),” AMl,*’ and PM3 (Ref. 29) Hamiltonians with these ab initio results of Hurst, Dupuis, and Clementi.’ Due to the semiempirical character of his calculations, he was able to work on oligomers up to C&H,, and to derive asymptotic values similar to those of Hurst, Dupuis, and Clementi. It is important to note here that the FF, the PEDM, as well as the CPHF methods give identical results though they are working differently because they are based on different techniques.
The previous studies have shown that the saturation pattern of the polarizability per unit cell is computationally difficult to reach. Indeed, for conjugated compounds, the strong delocalization is responsible for a very slow stabiliza- tion of these properties with respect to the number of atoms. Moreover, the asymptotic values obtained by using the ex- trapolation procedure proposed for example by Hurst, Du- puis, and Clement? depend on the number of points used in the fitting process. It is the reason for which it is necessary to
find methodologies which are suitable to provide the correct asymptotic values of response properties of optically active materials without using extrapolation schemes.
Only a few works have been devoted to the calculation of the polarizability of infinite chains. As mentioned before, the first calculations were performed using the Genkin- Mednis approach in the framework of the Hiickel approxi- mation. At the ab initio level, longitudinal electric polariza- bilities of infinite chains have already been computed using the same approach. Barbier analyzed the effect ofbond alter- nation in linear hydrogen chains.30 The longitudinal polar- izability of polyacetylene has also been computed.31 The dif- ficulty of differentiating the linear combination of atomic orbitals (LCAO) coefficients with respect to the wave vec- tor k, which are defined within a phase factor by the diagona- lization processes, was recently solved.32 This method was also applied for polyethylene and polysilane in minimal STO-3G and extended 4-3 1G basis sets in a way to associate the longitudinal polarizability to the topology of the elec- tronic band structure.32*33 However, it has not been stressed enough that the Genkin-Mednis approach corresponds to an uncoupled Hartree-Fock scheme [or summations over states (SOS) ] where the field-induced electron reorganiza- tion is not taken into consideration.
In this paper, a methodology based on the random- phase-approximation3b36 (RPA) method and its applica- tion to the calculation of the longitudinal asymptotic polar- izability per unit cell of an infinite polymer system is presented. As explained in the second part, it is not easy to apply the coupled Hartree-Fock (CHF) method to infinite systems. This is the reason why we have adopted the RPA method which is equivalent to the CHF method. This meth- od takes into account the electron reorganization induced by the application of an electric external field in contrast to the SOS technique. We show that the SOS (Genkin-Mednis) method may be considered as an approximation of the RPA one. We eventually study its ability and suitability by apply- ing the method to polyacetylene in the PPP approxima- tion. 17-19
II. EFFECT OF AN ELECTRIC EXTERNAL FIELD ON AN INFINITE PERIODIC SYSTEM A. The “unperturbed” periodic potential
The translational symmetry in quasi-one-dimensional systems (z is the chain direction) gives rise through B~oc~‘s theorem3’ to Bloch functions 4p (k,r) in a way to represent the one-electron wave functions. They consist in the multi- plication of a plane wave by a function up (k,r) which pre- sents the same periodicity as the lattice,
tip (k,r) = eikup (kr), (2) where k, which is a quasimomentum, is a continuous vari- able ranging from - r/u to V/U (a is the unit-cell length). This corresponds to the first Brillouin zone. So, an eigenstate corresponds to each k value in this interval. In the frame- work of the LCAO approximation, the n-labeled crystal or- bital Y,, (k,r) is written as a combination of Bloch functions q$ (kr),
Champagne, Fripiat, and AndrB: Polarizabilities of polymer chains 8331
J. Chem. Phys., Vol. 96, No. 11,l June 1992
*, (kr) = j) C,, (k)qhp (k,r), p=l
(3)
where a second index defines the band. Born-K&-man boundary conditions3* are commonly used to solve properly and efficiently the band-structure calculation. These cyclic boundary conditions impose that the (2N + 1) th cell is identical to the first one. The infinite cell-index summation defining the Bloch functions is then limited to a 2N + 1 sum- mation,
4,(k)= l ” ~~j=~~e~~aXp(I-P-joe.), (4)
where the first factor is the normalization factor, j is a cell index, andxp (r - P -jae, ) is the atomic function centered on thepth atom in thejth cell. e, is the unit vector in the z direction. These boundary conditions introduce 2N + 1 k- labeled states in each band with the k values restricted to a multiple of 25-/u (2N + 1) in the tist Brillouin zone. Usual- ly, in a way to obtain consistent results, N tends to infinity in order to numerically stabilize the lattice summations. The LCAO C,,, (k) coefficients as well as their associated ener- gies E, (k) are obtained by solving the generalized eigenval- ue problem,39
F(kWk) = S(k)C(k)dk), (5) where F(k) and S(k) are k-dependent Fock and overlap matrices.
B. The perturbed potential
By switching on a static electric external field parallel to the chain direction, the perturbed Hamiltonian loses its peri- odicity,
H(r) = H,(r) + eF,z, (6)
where e is the electron charge, F, is the applied field, H,, (r ) is the unperturbed periodic Hamiltonian, and z is the z com- ponent of the position operator (r). Bloch’s theorem then becomes invalid.
Churchill and Holmstrom40*41 studied the effects of the different boundary conditions on the free-electron wave functions when a field is turned on. Starting from the initial periodic wave functions, they obtained in this case the un- periodic Airy functions. They showed that, when the box boundary conditions [Y ( - L /2) = \I, (L /2) = 0 where L is the size of the box] are applied, the result is inconsistent if the field strength tends to zero and the position z tends to infinity, while the periodic boundary conditions (Born- K&man) cannot be imposed on the system. This last case would set one’s face against the second principle of thermo- dynamics because, physically, this situation is equivalent to one in which an electron reaches the end of the system at a potential - eFL and directly goes up to a situation where the potential is + eFL.
Since the Born-K&-man boundary conditions cannot be applied, a direct determination of the perturbed crystal orbi- tals and band energies is impossible. That is why such meth- ods as the numerical FF or the analytical CHF (or PEDM), which consists of the successive derivatives with respect to the field components of the generalized eigenvalue problem,
are not applicable in a band-structure formalism. The matrix consequence of the presence of the field is
the loss of hermiticity of the k-dependent Fock matrix F(k) due to the inclusion of the dipole moment matrix Z(k) gen- erated by the perturbation. These k-dependent matrix ele- ments are defined by
Z,(k) = 2 &kjaz Oj
w, (7) j= --N
where the cell-dependent elements may be reformulated as followed after the variable change “z = z’ +ju:”
By introducing this expression in the k-dependent formula and after inverting the sign in the summation, the nonhermi- ticity appears,
Z:(k) =Z,(k) +i+,(k).
8332 Champagne, Fripiat, and And&: Polarizabilities of polymer chains
C. Perturbational solutions
Since the methods to obtain the response properties, which need the calculation of the perturbed wave function or the derivatives of the unperturbed wave function with re- spect to the field strength are impossible, it is necessary to find procedures which directly compute the desired response functions. These techniques are based on perturbational ex- pansions. It is important to note here that the band struc- tures are evaluated at the Hartree-Fock level of approxima- tion which does not include the correlation error. Indeed, in this approximation, two electrons of different spins may oc- cupy the same spatial position. That is the reason why cor- rect polarizabilities will be obtained by considering two per- turbations: a one-electron perturbation due to the electric external field and a two-electron perturbation coming from the electron correlation correction. Propagator techniques are a suitable way to include the correlation correction in the computation of the response functions as will be presented in the next section.
iii. POLARIZATION PROPAGATOR METHODS TO OBTAIN THE ELECTRIC POLARiZABiLiTiES A. The RPA method applied to finite-size systems
When a system is submitted to an external time-depen- dent field oscillating at a frequency w,
F, (t) = F, cos wt, (10) where F, is the amplitude of the field, the exact frequency- dependent (through E which is equivalent w in the atomic units system) longitudinal polarizability3’36 is given in its spectral representation by
a,(E) = 2 2b9, -Eo)l(Olzlm)12
m (E,,, -Eo)‘-E2 ’ (11)
The summation runs over all the exact excited states Im) of energy E,,, . E. is the ground-state energy associated with the ground-state wave function (0) and (Olrlm) is a dipole integral between the ground state IO) and the excited
J. Chem. Phys., Vol. 96, No. iI,1 June 1992
Champagne, Fripiat, and AndrB: Polarizabilities of polymer chains 8333
In the case of closed-shell systems, using restricted Hartree- Fock (RHF) wave functions, the longitudinal polarizabili- ty, a,, takes the following form:
state 1 m) . This sum runs over an infinite set of excited states. The polarization propagator3”36 can provide a useful for- mula for the evaluation of the electric polarizabilities. In- deed, by using the superoperator resolvent form, the fre- quency-dependent polarizability is rewritten in an alternative form,
a,(E) = (O[z,(El+ HI-*zlO), (12) where [A$] = AB - BA is the commutator between the op- erators A and B and 1 and H are the superoperators4’ de- fined by the relations
L4 =A,
au(E) = 2W, -PI SE-j-A B (19)
where the different matrix elements are defined by
ts)ai,bj = sa*sQS w,i = (+I~>,
HA= [H$4]. (13) By introducing the binary product,43
(A I& = @[A +,B IO), (14) the longitudinal polarizability takes the following form:
a,(E) = (zl(El+ H)-‘z). (15) By combining with Lowdin’s inner projection techniqueM using a complete set of projection operators, the next form was obtained by Simons,43
where the chemists’ notation is used to defined the two-elec- tron integrals,
X&W2 14, (r2 1. (21) The indices now label the doubly occupied (unoccupied) molecular orbitals. For a static electric field, expression ( 18) becomes
a,(E) = (zlTt)(TtlEl+HITt)-‘(T+lz). (16) Since the operator r is a number-conserving operator the
complete set {rt} only has to contain the single, double, triple, etc. excitations or deexcitations. To make the previous equation workable, it is necessary to limit the operator mani- fold to some extent and to choose a starting wave function. The random-phase approximation (RPA) consists of limit- ing the operator manifold to single excitations and deexcita- tions and of using Hartree-Fock wave functions for the ground and excited states. This method, also called the first- order polarization propagator approximation, provides equivalent results to the FF or CHF procedures.3s Thus, it also takes into account the electron reorganizational effects due to the presence of an electric field. This approximation may be improved by introducing multiple excitations and also correlated wave functions in a way to evaluate the polar- izability accurately through a higher order in the perturba- tion expansion. For example, in the second-order polariza- tion propagator approximation, the results are consistent with the second order in the treatment of the electron corre- lation.36 This fact characterizes the great power of propaga- tor techniques which may be adapted to the accuracy of the desired values.
a, =2(P,-P)(;: 3-y _pp), (22)
or, in a more compact form, since all matrix elements are real,
arr = 4P( A - B) - ‘P. (23) To solve this equation, an intermediate step consists of
the inversion of the problem (A - B) - ‘P = X into the lin- ear equations systems (A - B) X = P.
B. The RPA method applied to infinite systems
The polymeric electronic states are characterized by two labels: the band index n and the quasimomentum k. So, an excitation (or deexcitation) is represented by four in- dices. You are, however, left with one k label because the z- dipole integrals (z is the chain direction) between an occu- pied and unoccupied Bloch functions are only nonzero for k = k t,45
The operator manifold for RPA is then given by CT”) = {Q ‘,QI = Caf,i,UiaI, (17)
where at (a) is a creation (annihilation) operator. The la- bels u,b,... (id,...) correspond to particle (hole) spin orbitals. By partitioning the operator space into Q and Q +, the polar- ization propagator takes the form a,(E) = [(zlQt)(4Q)l
(da(k’)lZl$i(k)> =akktPai(k)* (24) This method thus requires the evaluation of the dipole
integrals between crystal orbitals. As mentioned by Callaway, it is evident that there will be difficulties asso- ciated with the calculations of these dipole integrals because, for sufficiently large distances, the perturbation becomes ar- bitrarily large, no matter how weak the field is. Ladik47 has recently shown that, despites the unbounded character of the operator position, z, all the dipole matrix elements are finite. According to Blount’s procedure,45 the dipole inte- grals may be rewritten in the following form:
= islai(k), (25) where the dipole transition strengths fIOi (k) are defined by a
J. Chem. Phys., Vol. 96, No. 11,l June 1992
8334 Champagne, Fripiat, and And&: Polarizabilities of polymer chains
space integration over a unit cell. Barbier, Delhalle, and An- drC4* provided an analytical expression of Qai ( k) , and more recently, we published a better numerical form33 for compu- tation.
By using the transformation of the dipole integrals into the dipole transition strengths [ Eq. (25) ] and the anti-Her- mitian character of these latter integrals,33 the frequency- independent polarization propagator for closed-shell infinite systems may be written as
a, = 2u-l*4,4 f*) - I(;*), (26)
where the asterisk indicates the complex conjugated values because of the complex nature of the Bloch functions. The matrix elements are defined by
Expression (26) is similar to the previously described mo- lecular one [ Eq. (22) 1, except there is an infinite number (2N + l,N+ CO ) of k values or of k states in one band. Thus, the matrices A, B, and s1 are of infinite dimension. It is wor- thy to note that the Kronecker delta Sk,, corresponds to a continuous Dirac delta function except for a multiplicative factor. This factor depends on the number of k states in the first Brillouin zone,
s 29.r ,,,++-----S(k - k’). d2N+ 1)
(28)
The first step is to solve the linear equations system obtained by inverting the right part of the polarization propagator,
AX+BY=n, B*X + A*Y = a*. (29)
It is important to keep in mind that there exist an equation for each triplet formed by a k value and two band indices representing an excitation. Thus the left-hand part of these secular equations contain twice the number of all possible excitations. The infinite sum over k may then become an integration over the first Brillouin zone,
In this preliminary work, trapezoidal quadrature is used to obtain X(k) and Y(k) . As N tends to infinity, the divergent character of the multiplicative factor in-front is compensat- ed by the normalization factor of the crystal orbitals LnmTI.
The second step consists of multiplying the two follow- ing infinite vectors to obtain the longitudinal polarizability of the infinite system:
a, = W*(k),fi(k)l($$). (33)
The summation over k is replaced by an integration in the first Brillouin zone which straightforwardly provides the longitudinal polarizability per unit cell:
(30)
(31)
(32)
I
au (2N+ 1) =;T”FJy;,a [f$(k)X,(k)
+ fi,(k)Y,(k)]dk. (34) As in the case of the SOS method,32 the RPA expression of the longitudinal polarizability per unit cell can also be parti- tioned into the contributions of each occupied band,
a, =Tf [“~SJ~~,, [‘$(k)X,(k) (2Nt 1) j
+ fibj(k)Ybj(k)]dk)
(35)
J. Chem. Phys., Vol. 96, No. II,1 June 1992
By making the approximation where matrix B is zero and A is restricted to A,, , the matrices become diagonal, ev- ery coupling between the crystal orbitals disappears, and the polarizability reads as
~=~“~~~~,a [ E 2N+ 1
;;)fk$] dk. (36) - B I
In this formula, there is no coupling between different verti- cal transitions (k stays constant) whereas such coupling is present through the matrices A, and B. This coupling and thus the matrices A, and B are responsible for the field- induced electron reorganizational effects.
IV. APPLICATION TO MODEL POLYACETYLENE CHAINS
In this preliminary work, the potentials of the method are discussed in the framework of the PPP method. “-19 The chosen model prototype systems are polyacetylene chains where only the r structure is taken into account. The longi- tudinal polarizabilities of finite increasing chains are firstly computed to check the behavior of the evolution of their values per unit cell. Then, by using our RPA method applied to infinite systems, the asymptotic longitudinal polarizabili- ty per unit cell is obtained and compared with the value of the larger oligomers. For the sake of comparison, the SOS- uncoupled analogous values are also computed. Standard geometrical parameters are taken. A double (single) bond is 1.35 ( 1.46) A long and all angles are of 120”.
The zero differential overlap (ZDO) approximation present in the PPP method leads to an orthogonal atomic basis set and to the following expressions for the Fock diag- onal and off-diagonal matrix elements for the finite systems:
where Wpp is the energy of an electron in the 2p, orbital of a carbon atom ( - 11.28 eV), Ppp is the r-electronic charge on the carbonp, and P, is the bond index between the atomsp and q. For the infinite systems, the k-dependent Fock matrix elements read as
F,(k) = 2 eikjaFz, j= -N
(38)
where the cell-dependent Fock elements are also defined for the diagonal and off-diagonal cases,
where the sum over r runs over all the atomic functions. The superscript indices refer to the unit cell containing the atom- ic orbital indexed in the subscript. The prime indicates the exclusion from the summation of the term wherej = 0 and r = p simultaneously.
(39) TABLE I. Bond orders for C&H,, according to the parametrization.
Parker Tavan
Double bond 0.9595 0.8844 Single bond 0.2023 0.3450
Among the various PPP parametrizations, Ohno’s for- mula49 is chosen in this work for the Coulombic repulsion integral,
(ppjqq) = 14.397( 1.634 81 + R&) -“* eV. (4-o) In the last expression, the distances R,, between the atomsp and q are given in A. For the resonance integrals (p), two versions are used in a way to show their effects: the first fl expression was optimized by Pariser and Parr’* to give the best lowest singly excited states of ethylene and benzene,
ppq = - 6442e- 5’6a64Rw eV. (41) In another parametrization, /? is only nonzero for neighbor- ing atoms and is given by a linear equation used by Tavan4* to provide a good agreement between experimental and theoretical excitation energies of polyene chains. Formulas of this type are used by Soos and de Melo,
#?, = - 2.6 + 3.21(R, - 1.397) eV. (42) The difference between the two p parametrizations lies
in the sharper decreasing character of the Pariser formula. This leads to a more important electron-density alternation as shown in Table I by the bond orders of the oligomer that contains 15 unit cells.
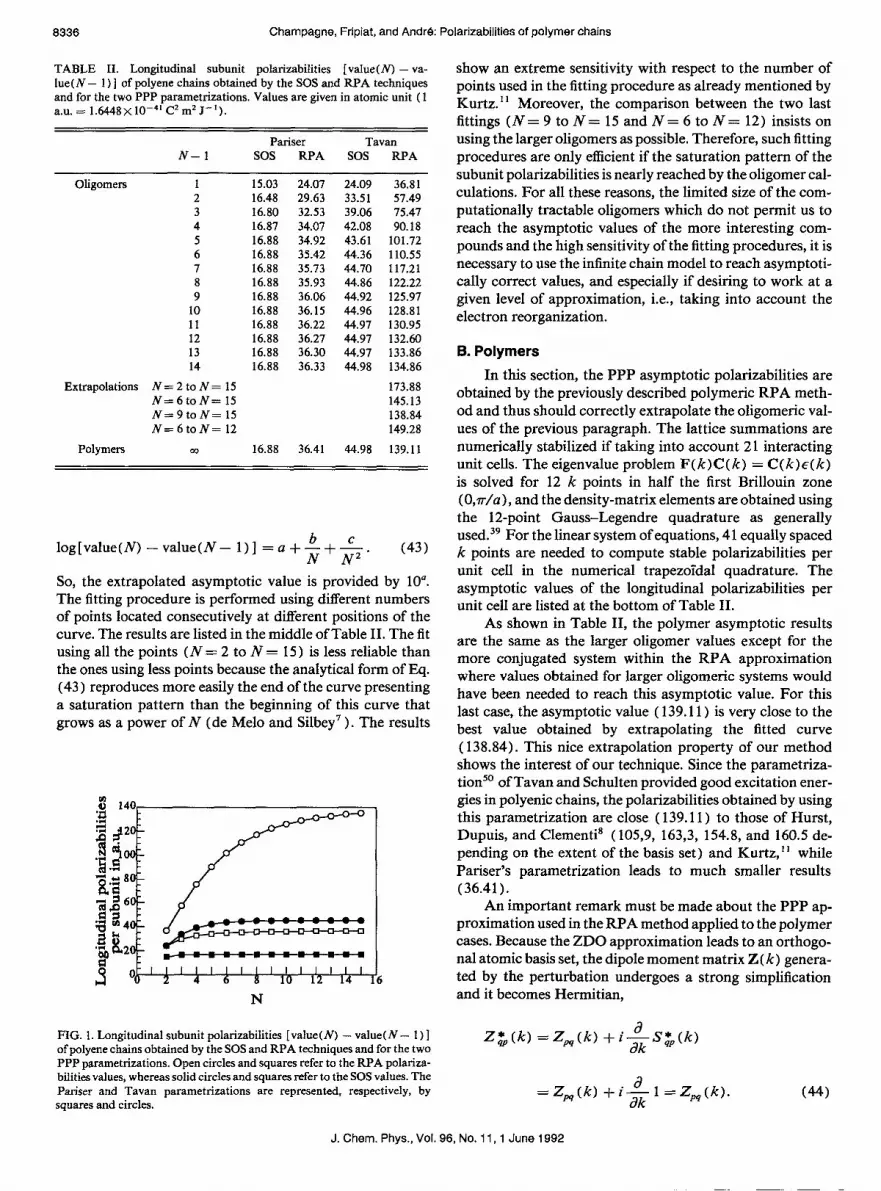
A. Oligomers Table II and Fig. 1 show the longitudinal subunit polari-
zabilities of the size increasing oligomers (Nis the number of unit cells composed of two carbon atoms) within the two parametrizations by using the SOS (uncoupled) and RPA (coupled) techniques previously described. These are repre- sented in Fig. 1. Kurtz I1 has shown that the difference be- tween the polarizabilities obtained with the N and N - 1 systems describes the appearance of a saturation pattern bet- ter than the more frequently used polarizability per unit cell (a,/N).
As expected, the electron-density alternation affects the polarizabilities which are larger for the less-altemant hydro- carbon chains. The field-induced electron reorganization through the RPA leads to larger values than the SOS ones. Moreover, the more regular the chain (the more conjugated the chain), the larger the RPA/SOS ratio is. Thus the SOS method gives poor results for the highly conjugated systems, which are the most interesting ones in nonlinear optics. This implies that it is necessary to use methods which at least take into account the electron reorganization, i.e., the RPA or equivaIent methods. Figure 1 shows also that the asymptotic value is not reached for the well-conjugated system using the RPA method.
In order to find this asymptotic value, the procedure of Hurst, Dupuis, and Andre’ and Kurtz” is followed and a least-squares fit is performed on the equation,
Champagne, Fripiat, and And&: Polarizabilities of polymer chains 8335
J. Chem. Phys., Vol. 98, No. 11,l June 1992
TABLE II. Longitudinal subunit polar&abilities [value(N) - va- lue( N - 1) ] of polyene chains obtained by the SOS and RPA techniques and for the two PPP parametrizations. Values are given in atomic unit (1 a.u. = 1.6448~ 10m4’ C!’ m2 J-I).
Extrapolations N = 2 to N = 15 173.88 N=6toN=l5 145.13 N=9toN= 15 138.84 N=6toN= 12 149.28
Polymers 03 16.88 36.41 44.98 139.11
8336 Champagne, Fripiat, and And&: Polarizabilities of polymer chains
show an extreme sensitivity with respect to the number of points used in the fitting procedure as already mentioned by Kurtz. ’ ’ Moreover, the comparison between the two last fittings (N= 9 to N= 15 and N= 6 to N= 12) insists on using the larger oligomers as possible. Therefore, such fitting procedures are only efficient if the saturation pattern of the subunit polarizabilities is nearly reached by the oligomer cal- culations. For all these reasons, the limited size of the com- putationally tractable oligomers which do not permit us to reach the asymptotic values of the more interesting com- pounds and the high sensitivity of the fitting procedures, it is necessary to use the infinite chain model to reach asymptoti- cally correct values, and especially if desiring to work at a given level of approximation, i.e., taking into account the electron reorganization.
6. Polymers
log[value(N) -value(N- l)] =a+$+$. (43)
So, the extrapolated asymptotic value is provided by 10”. The fitting procedure is performed using different numbers of points located consecutively at different positions of the curve. The results are listed in the middle of Table II. The fit using all the points (N = 2 to N = 15 ) is less reliable than the ones using less points because the analytical form of Eq. (43) reproduces more easily the end of the curve presenting a saturation pattern than the beginning of this curve that grows as a power of N (de Melo and Silbey’ ). The results
In this section, the PPP asymptotic polarizabilities are obtained by the previously described polymeric RPA meth- od and thus should correctly extrapolate the oligomeric val- ues of the previous paragraph. The lattice summations are numerically stabilized if taking into account 2 1 interacting unit cells. The eigenvalue problem F( k)C( k) = C( k)e( k) is solved for 12 k points in half the first Brillouin zone (O,?r/u>, and the density-matrix elements are obtained using the 1Zpoint Gauss-Legendre quadrature as generally used.39 For the linear system of equations, 41 equally spaced k points are needed to compute stable polarizabilities per unit cell in the numerical trapezoidal quadrature. The asymptotic values of the longitudinal polarizabilities per unit cell are listed at the bottom of Table II.
As shown in Table II, the polymer asymptotic results are the same as the larger oligomer values except for the more conjugated system within the RPA approximation where values obtained for larger oligomeric systems would have been needed to reach this asymptotic value. For this last case, the asymptotic value ( 139.11) is very close to the best value obtained by extrapolating the fitted curve (138.84). This nice extrapolation property of our method shows the interest of our technique. Since the parametriza- tion” of Tavan and Schulten provided good excitation ener- gies in polyenic chains, the polarizabilities obtained by using this parametrization are close ( 139.11) to those of Hurst, Dupuis, and Clementi’ ( 105,9, 163,3, 154.8, and 160.5 de- pending on the extent of the basis set) and Kurtz,” while Pariser’s parametrization leads to much smaller results (36.41).
An important remark must be made about the PPP ap- proximation used in the RPA method applied to the polymer cases. Because the ZDO approximation leads to an orthogo- nal atomic basis set, the dipole moment matrix Z(k) genera- ted by the perturbation undergoes a strong simplification and it becomes Hermitian,
FIG. 1. Longitudinal subunit polarizabilities [value(N) - value( N - 1) ] of polyene chains obtained by the SOS and RPA techniques and for the two PPP parametrixations. Open circles and squares refer to the RPA polarixa- bilities values, whereas solid circles and squares refer to the SOS values. The Parker and Tavan parametrixations are represented, respectively, by squares and circles.
=Z,(k) ++ =Z,(k). (4)
J. Chem. Phys., Vol. 96, No. 11,l June 1992
Champagne, Fripiat, and Andr6: Polarizabilities of polymer chains 8337
This simplification allows the determination of field-per- turbed crystal orbitals and band energies because the Fock matrix stays Hermitian. In this case, the methods such as the FF, CHF or PEDM ones are suitable to obtain the asympto- tic value. We have, however, described the general RPA pro- cedure to obtain the asymptotic polarizabilities without tak- ing into account such an approximation. Work is in progress to implement this method at the ab initio level.
V. SUMMARY, CONCLUSIONS, AND OUTLOOK
We have formulated a general method to compute the asymptotic longitudinal polarizabilities of polymer com- pounds which takes into account the field-induced electron reorganizational effects. This method consists in the applica- tion of the RPA technique to the infinite compounds. The previously used SOS or uncoupled Hartree-Fock scheme is a poor approximation of this new method since it does not take into account the reorganizational effects.
To test its ability and suitability, the method was applied to model prototype P systems of polyacetylene chains in the framework of the PPP approximation. First, the longitudi- nal polarizabilities per subunit were obtained for size-in- creasing oligomers. The RPA/SOS ratio was shown to be larger for the more conjugated compounds and thus the RPA more adapted to compute the polarizabilities of the more interesting compounds. The slow convergence of the polarizability per subunit of the more conjugated com- pounds within the RPA as well as the difficulties related to the application of fitting procedures to extrapolate the oli- gomer results demands techniques which compute directly the asymptotic values. The asymptotic values computed by our method coincide with the subunit values of the larger oligomers or provide the desired asymptotic values, very close to the best extrapolated values, when the system was not sufficiently large to reach the saturation pattern.
The next step is to implement our method at the ab initio level where computation-time and disk-space problems would raise our attention especially on computational de- tails as the lattice sum truncations and the number of k points for the integrations.
ACKNOWLEDGMENTS The authors would like to thank Dr. D. H. Mosley for
his comments. One of us (B.C.) thanks the Belgian National Fund for Scientific Research (FNRS) for his Research As- sistant position. All calculations reported here have been performed on the Namur-Scientific Computing Facility, a result of a cooperation between the FNRS, IBM-Belgium, and the Facultes Universitaires Notre-Dame de la Paix.
’ Nonlinear Optical Properties of Organic Molecules and Crystals, edited by D. S. Chemla and J. Zyss (Academic, New York, 1987), Vols. 1 and 2.
‘V. P. Bodart, J. Delhalle, J. M. Andre, and J. Zyss, in Polydiacetylenes: Synthesis, Structure and Electronic Properties, edited by D. Bloor and R. R. Chance (Martinus Nyhoff, Dordrecht, 1985), p. 125.
‘H. F. Hameka, J. Chem. Phys. 67,2935 (1977).
‘0. Zamani-Khamiri and H. F. Hameka, J. Chem. Phys. 71,1607 ( 1979). ‘M. G. Papadopoulos, J. Waite, and C. A. Nicolaides, J. Chem. Phys. 77,
2527 (1982). 6 V. P. Bodart, J. Delhalle, J. M. Andre, and J. Zyss, Can. J. Chem. 63, 163 1
(1985). ‘C. P. de Melo and R. Silbey, J. Chem. Phys. 88,2558 (1988). sG. J. B. Hurst, M. Dupuis, and E. Clementi, J. Chem. Phys. 89, 385
(1988). 9B. Kirtman, Chem. Phys. Lett. 143,81 (1988). ‘OZ. G. Soos and G. W. Hayden, Phys. Rev. B 40,308l (1989). “H. A. Kurtz, Int. .I. Quantum Chem. Quantum Chem. Symp. 24, 791
(1990). I2 B. Champagne and J. M. Andre, Int. J. Quantum Chem. Quantum Chem.
Symp. 24,859 (1990). I3 V. M. Genkin and P. M. Mednis, Zh. Eksp. Teor. Fiz. 54, 1137 ( 1968)
[Sov. Phys. JETP 27,609 (1968)]. “C. Cojan, G. P. Agrawal, and C. Flytzanis, Phys. Rev. B 15,909 (1977). 15H. F. Hameka and E. N. Svendsen, Int. J. Quantum Chem. 10, 249
(1976). 16H. F. Hameka and E. N. Svendsen, Int. J. Quantum Chem. 11, 129
(1977). “R. Pariser, J. Chem. Phys. 21, 568 (1953). I8 R. Pariser and R. G. Parr, J. Chem. Phys. 21,767 (1953). I9 J. A. Pople, Trans. Faraday Sot. 49, 1375 ( 1953). ‘OR. McWeeny, Phys Rev. 126, 1028 (1962). “J. A. Pople and G. A. Segal, J. Chem. Phys. 44,3289 ( 1966). **J A. Pople, D. L. Beveridge, and P. A. Dolbosh, J. Chem. Phys. 47,2026
(1967). 23 H. D. Cohen and C. C. J. Roothaan, J. Chem. Phys. 43,534 (1965). “W. J. Hehre, R. F. Stewart, and J. A. Pople, J. Chem. Phys. 51, 2657
(1969). ‘sR. Ditchfield, W. J. Hehre, and J. A. Pople, J. Chem. Phys. 54, 724
(1971). 26A. Dalgamo, Adv. Phys. 11,281 (1962). “M. J. Dewar and W. Thiel, J. Am. Chem. Sot. 99,4899 ( 1977). ‘*M. J. Dewar, E. G. Zoebisch, E. F. Healy, and J. J. P. Stewart, J. Am.
Chem. Sot. 107,3902 (1985). *9 J. J. P. Stewart, J. Comput. Chem. 10,209 ( 1989). s”C. Barbier, Chem. Phys. Lett. 142,53 (1987). 3’ C. Barbier, J. Delhalle, and J. M. Andre, in Nonlinear Optical Properties
ofPolymers, edited by A. J. Heeger, J. Orenstein, and D. R. Ulrich (Mate- rials Research Society, Pittsburgh, 1988), Vol. 109, p. 143.
“B. Champagne and J. M. Andre, Int. J. Quantum Chem. (to be pub- lished)
33 J. M. Andre and B. Champagne, in Organic Moleculesfor Nonlinear Op- ticsand Photonics, edited by J. Messier and F. Kajzar (Kluwer Academic, Dordrecht, 1991), p. 1.
34J. Oddershede, Adv. Quantum Chem. 11,257 (1978). 35P Jdrgensen and J. Simons, Second Quantization-Based Methods in
Quantum Chemistry (Academic, New York, 198 1) . 36 J. Gddershede, P. Jdrgensen, and D. L. Yeager, Comput. Phys. Rep. 2,33
(1984). 37F. Bloch, Z. Phys. 52,555 (1928). 38 Eleczronic Structure of Polymers an Molecular Crystals, edited by J. M.
Andre and J. Ladik (Plenum, London, 1974). 39 J. M. Andre, J. Delhalle, and J. L. Bredas, Quantum Chemistry Aided
Design of Organic Polymers for Molecular Electronics (World Scientific, London, 1991), Chaps. 1 and 2.
“OJ. N. Churchill and F. E. Holmstrom, Am. J. Phys. 50,848 ( 1982). 4’ J. N. Churchill and F. E. Holmstrom, Physica B 123, 1 ( 1983). 42B. T. Pickup and 0. Goscinski, Mol. Phys. 26, 1013 (1973). 43P. 0. Lowdin, Phys. Rev. 139, A357 (1965). 44 J. Simons, J. Chem. Phys. 64,454l ( 1976). ‘sE. I. Blount, in Solid States Physics, edited by F. Seitz and D. Tumbull
(Academic, New York, 1962), Vol. 13, p. 305. &J. Callaway, Energy Band Theory (Academic, New York, 1964), p. 277. 47J. Ladik, J. Mol. Struct. (Theochem) 206, 39 (1990). “C. Barbier, J. Delhalle, and J. M. Andre, J. Mol. Struct. (Theochem) 188,
299 (1989). ‘9K. Ohno, Theor. Chim. Acta 2,219 (1964). 5oR. Tavan and K. Schulten, J. Chem. Phys. 70,5407 (1979).