Ethylene Homopolymerization with P, 0-Chelated Nickel Catalysts U. KLABUNDE,* R. MULHAUF'T,t T. HERSKOVITZ, A. H. JANOWICZ, J. CALABRESE, and S. D. I'M'EL, **CentralResearch & Development Department, Experimental Station, E. I. du Pont de Nemours & Company, Wilmington,Delaware 19898 Synopsis Abstraction of phosphine from the nickel(I1) P, 0-chelated complexes, Ni[Ph, PCH=C(Ph)O] (Ph)(PPh,), and related species converts them from olefin oligomerization to olefin polymeriza- tion catalysts. Phosphine acceptors such as Rh(acetylacetonate)(C2H4)2 or Ni(l,5-cyclooctadiene), are most effective. Alternatively, nickel complexes-inwhich the phosphine ligand is replaced with weakly coordinated pyridine can be prepared. These active, homogeneous catalysts can be tuned to give either low or high molecular weight, linear low or high density polyethylene. Depending on the diluent, the same catalytic complex can be used as heterogeneous or homogeneous catalyst. They are tolerant of oxygenated, hydroxylic, or polar molecules that would poison normal early transition metal-based Ziegler-Natta catalysts. In fact, the polymerizations can be run in solvents such as ethanol or acetone, but hydrocarbon solvents are preferred. INTRODUCTION The polyolefin industry relies upon Ziegler-Natta, chromium oxide, and other catalysts based upon early transition metals.' Although the array of catalysts available offers many different approaches to the manufacture of polyolefins having a variety of physical properties,2 these catalysts are all extremely susceptible to deactivation by a range of poisons. Primary among these poisons are traces of oxygen, carbon monoxide, and water that can make their way into manufacturing facilities or laboratory experiments in a variety of ways. Other oxygen donors such as ethers, alcohols, or ketones can be poisons for these catalysts. As a result, the industry must carefully purify the ethylene and solvents used for polyolefin manufacture. This sensitivity to oxygenated species also precludes copolymerization of ethylene with polar monomers such as those containing ester or nitrile functionality. A catalyst that could accomplish coordination polymerization of ethylene with polar comonomers under moderate pressures is clearly of interest. Our search for new catalyst systems resistant to deactivation by oxygenated species focused on the late transition metals because they are less oxophilic than the early metals. Shell3 has developed a nickel-based ethylene oligomer- ization catalyst that yields higher 1-olefins,but their patents4 and the patents *Authors to whom correspondence should be addressed. +Current address: Ciba Geigy AG, Forschungszentnun KA/Marly, CH-1701 Fribourg, Switzer- *Author to whom requests for structural details should be addressed. land. Journal of Polymer Science: Part A: Polymer Chemistry, Vol. 25, 1989-2003 (1987) 0 1987 John Wiley & Sons, Inc. CCC 0360-3676/87/071989-15$04.00
Transcript
Ethylene Homopolymerization with P, 0-Chelated Nickel Catalysts
U. KLABUNDE,* R. MULHAUF'T,t T. HERSKOVITZ, A. H. JANOWICZ, J. CALABRESE, and S. D. I'M'EL, **Central Research & Development Department, Experimental Station, E. I. du Pont de
Nemours & Company, Wilmington, Delaware 19898
Synopsis
Abstraction of phosphine from the nickel(I1) P, 0-chelated complexes, Ni[Ph, PCH=C(Ph)O] (Ph)(PPh,), and related species converts them from olefin oligomerization to olefin polymeriza- tion catalysts. Phosphine acceptors such as Rh(acetylacetonate)(C2H4)2 or Ni(l,5-cyclooctadiene), are most effective. Alternatively, nickel complexes-in which the phosphine ligand is replaced with weakly coordinated pyridine can be prepared. These active, homogeneous catalysts can be tuned to give either low or high molecular weight, linear low or high density polyethylene. Depending on the diluent, the same catalytic complex can be used as heterogeneous or homogeneous catalyst. They are tolerant of oxygenated, hydroxylic, or polar molecules that would poison normal early transition metal-based Ziegler-Natta catalysts. In fact, the polymerizations can be run in solvents such as ethanol or acetone, but hydrocarbon solvents are preferred.
INTRODUCTION
The polyolefin industry relies upon Ziegler-Natta, chromium oxide, and other catalysts based upon early transition metals.' Although the array of catalysts available offers many different approaches to the manufacture of polyolefins having a variety of physical properties,2 these catalysts are all extremely susceptible to deactivation by a range of poisons. Primary among these poisons are traces of oxygen, carbon monoxide, and water that can make their way into manufacturing facilities or laboratory experiments in a variety of ways. Other oxygen donors such as ethers, alcohols, or ketones can be poisons for these catalysts. As a result, the industry must carefully purify the ethylene and solvents used for polyolefin manufacture. This sensitivity to oxygenated species also precludes copolymerization of ethylene with polar monomers such as those containing ester or nitrile functionality. A catalyst that could accomplish coordination polymerization of ethylene with polar comonomers under moderate pressures is clearly of interest.
Our search for new catalyst systems resistant to deactivation by oxygenated species focused on the late transition metals because they are less oxophilic than the early metals. Shell3 has developed a nickel-based ethylene oligomer- ization catalyst that yields higher 1-olefins, but their patents4 and the patents
*Authors to whom correspondence should be addressed. +Current address: Ciba Geigy AG, Forschungszentnun KA/Marly, CH-1701 Fribourg, Switzer-
*Author to whom requests for structural details should be addressed. land.
Journal of Polymer Science: Part A: Polymer Chemistry, Vol. 25, 1989-2003 (1987) 0 1987 John Wiley & Sons, Inc. CCC 0360-3676/87/071989-15$04.00
1990 KLABUNDE ET AL.
of others working in the area6 do not disclose high molecular weight polyethyl- ene or copolymerizations. Bayer AG reports the polymerization of ethylene with catalysts derived from nickel and phosphorus y l i d ~ . ~ Keim' and others' have continued to study the nickel systems in detail, and Keimg has reported an interesting catalyst system that provides 1-olefins in toluene but higher molecular weight polymers in h e m e under the same conditions. These reports prompted us to investigate the nickel system more carefully. In a series of papers, we wi l l discuss our work on o l e h polymerization with these nickel catalysts. In the first paper, we discuss the synthesis of the catalysts, their application to ethylene homopolymerization, and the factors that affect catalyst activity and polymer properties. In subsequent papers we wil l discuss the synthesis of a unique U D P E and polar-functionalized LLDPE. We will also discuss the molecular reactivity of the catalysts.
EXPERIMENTAL
Many of the required operations involve moderately moisture- or air- sensitive materials and were carried out in the prepurified nitrogen atmo- sphere of a Vacuum Atmospheres drybox. Operations outside the drybox were carried out using standard Schlenk techniques.'O Solvents were dried by standard techniques." Trimethylaluminum was obtained from Ethyl Corporation and was used without further purification. Ni(l,5-cyclooctadiene), was prepared by the method of Schunn." Rh(2,4-pentanedionate)(ethylene), was prepared by the method of Cramer.', Nickel complexes of the type RNi(R,PCR=CRO)(PR,) were prepared by literature methods,' and their structures are listed in Table I.
TABLE I Catalyst Structures
Catalyst number L
1 2 3 4 5 6 7 8 9 108 l l b
H Ph Ph Ph Ph SO, Na H S0,Na Ph H H
Ph OCH, Ph OCH, Ph Ph Ph Ph Ph Ph Ph
PPh, PEt , PEt , PPh, PPh, PPh, NC5H5 NC5H5 N C P 5 h e r
aDimer with bridging 0 in place of L. bTwo chelating ligand systems bridged at 0 by dimethyl-aluminum. Methyl group replaces Ph.
ETHYLENE HOMOPOLYMERIZATION 1991
Catalyst Synthesis
Synthesis of Ni(Ph,PCH=C(Ph)O),
A toluene solution (300 mL) of benzoylmethylene triphenylphosphorane (7.1 g, 18.7 mmol) and bis(l,5-~yclooctadiene)nickel (2.6 g, 9.5 mmol) was reacted with an excess of methyl methacrylate at room temperature for 19 h. The solution was filtered to remove traces of nickel metal, and the toluene was removed under reduced pressure. The solid residue was recrystallized from methylene chloride/ethanol by partial removal of the methylene chloride under reduced pressure, giving 3.8 g of deep orange crystals. Further reduction of the filtrate gave an additional 0.8 g for a combined yield of 75%. The complex is air-stable both in solution and in the solid state.
Synthesis of MeNi[(Ph,PCH=C(Ph)O),AlMeJ
A solution of 1 (see Results section; 1.10 g, 1.68 mmol) in toluene (75 mL) was reacted with a slight excess of trimethylaluminum (0.18 g, 2.50 mmol). A homogeneous, honey-brown solution was obtained. Ether (5 mL) and hexane (20 mL) were added, and the mixture was allowed to stand. The aluminum- bridged complex precipitated as an orange solid that was collected and washed with hexane. Yield was 1.1 g (90%).
Synthesis of PhNi(Ph, PCH =C(Ph)O)(NC,HJ and Related Complexes
Benzoylmethylene triphenylphosphorane (3.20 g, 8.42 mmol), bis(l,5- cyc1ooctadiene)nickel (2.31 g, 8.42 mmol), and an excess of pyridine (10.8 g) were dissolved in toluene (200 mL). The mixture was heated to 50°, allowed to cool to room temperature, and stirred for 16 h. After addition of diatomaceous earth, the solution was filtered to remove a small amount of nickel metal. Volatiles were removed under reduced pressure. Filtration of a hexane suspen- sion gave 3.8 g of a yellow solid that was recrystallized from warm toluene to which hexane was added.
The complex PhNi(Ph,PC(SO,Na)=C(Ph)O)(NC,H,) was prepared simi- larly starting from Ph,P=C(SO,Na)C(O)Ph (4.8 g). The yield was 4.9 g.
The complex PhNi(Ph,PC(SO,Na)=C(Ph)O)(p-MeC,H,N) was prepared similarly starting from Ph,P=C(SO,Na)C(O)Ph and 4-picoline.
The complex PhNi(Ph, PCPh=C(Ph)O)(Py) was prepared similarly starting from Ph,P=CPhC(O)Ph (5.05 g). The yield was 5.3 g.
Preparation of [PhNi(Ph,PCH =C(Ph)-p-O)]2
A freshly prepared benzene solution (70 mL) of PhNi(Ph,CH=C(Ph)O) (PEt,) (2.70 g, 4.87 -01) was reacted with the phosphine acceptor compound Rh(acac)(C,H,), (0.68 g, 2.64 mmol) (acac = 2,4-pentanedionato). The solu- tion was promptly filtered to remove a small quantity of insolubles. On standing, 0.69 g of honey-brown crystals precipitated. After isolation by vacuum filtration, the crystals were washed with a small amount of benzene. The filtrate was h&ed to 60°, 130 mL of benzene and 50 mL of hexane were
1992 KLABUNDE ET AL.
added, and the mixture was allowed to stand at room temperature for 72 h. An additional 0.25 g of product was isolated for a combined yield of 44%.
Ethylene Polymerization by the Ni Catalysts
General Proceohre
A 500 mL Fisher-Porter pressure bottle was charged under nitrogen with 50-200 mg of the polymerization catalyst or catalyst precursor and ligand acceptor. The head assembly of the pressure reactor was of a Du Pont design commercialized by Fisher-Porter. It was equipped with a dip tube for removal of small liquid samples for gas chromatographic analysis, an inlet valve for evacuation and charging of gases, a pressure gauge, a thermocouple, and an injection port similar to those found on gas chromatographs. Early experi- ments were stirred magnetically, but later experiments were stirred mechani- cally at 500 rpm controlled by a Servodyne speed controller. The bottle was briefly evacuated and then charged with 50-75 psig (345 or 518 kPa) of ethylene for 2 min. The valve to the ethylene was closed, the bottle was partially immersed in a heated oil bath, and the solution was stirred. There was an initial pressure rise due to the increase in temperature. As the pressure fell back to the,set pressure, the valve to ethylene was reopened, and the pressure was maintained for the duration of the polymerization. Samples for GC analysis were typically removed every 30 min. Polymerizations were typically run for 2-4 h, but on occasion were run for much shorter or longer periods of time. The polymerizations were terminated by release of the ethylene pressure. A final small liquid sample was taken for chromatographic analysis to determine oligomer content. The contents of the bottle were transferred in air to a 100 mL beaker containing methanol (600 mL). Catalyst residues were readily removed by methanol wash, and white polymers were obtained. The suspension was stirred until the polymer was white. It was collected by filtration, washed with several portions of methanol, and dried at room temperature under vacuum.
The details of individual polymerizations are presented in Table 11. Vari- ables include the structure and quantity of catalyst or catalyst mixture, the solvent, the polymerization temperature, and the length of polymerization. The presence of low molecular weight oligomers is noted for those cases where they were analyzed. The higher pressure runs indicated in Table I1 were carried out in a 1 L stainless steel autoclave with internal cooling for temperature control. The constant temperature runs, indicated by footnote (g) in Table 11, were done in the standard apparatus that had been modified to include a high efficiency cooling coil immersed in the polymerization medium. The results of a series of polymerizations with added polar molecules are presented in Table 111.
Ethylene Polymerization and Isolation of Spent Catalyst
A polymerization was carried out in the glass reactor as described above, but rather than the standard 0.1 g of catalyst, 1.0 g of PhNi[Ph,CPh=CPh(0)](NC5H5) was employed. After ethylene uptake ceased, 27 g of polymer were isolated. The solvent was evaporated, and the
ETHYLENE HOMOPOLYMERIZATION 1993
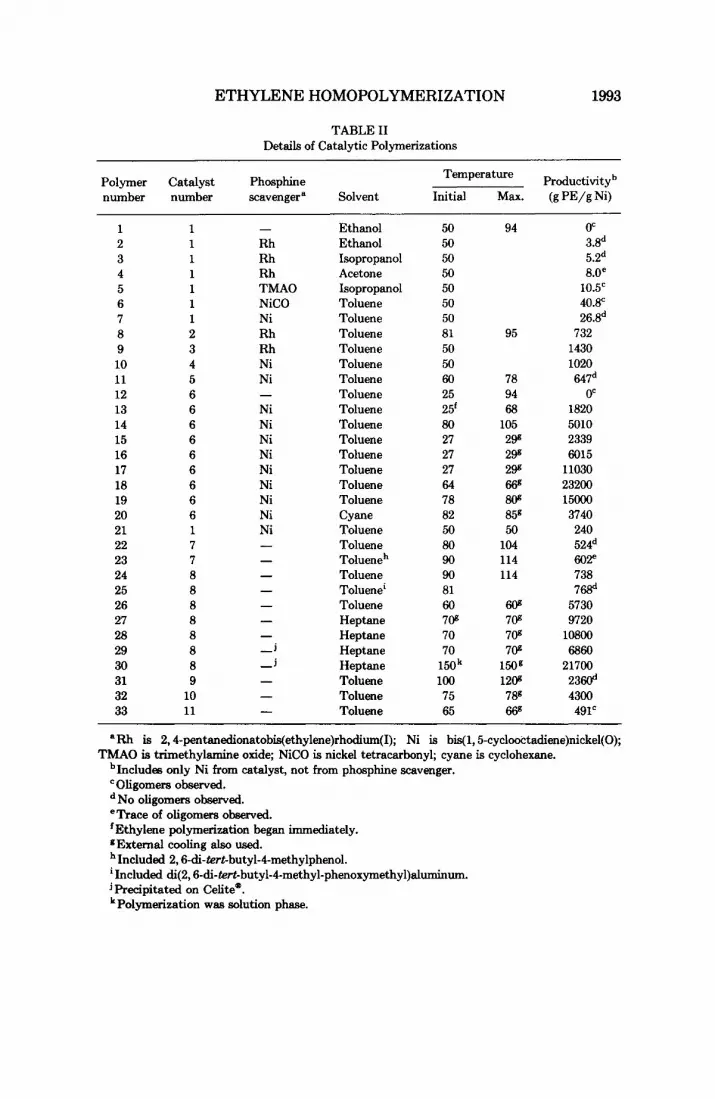
TABLE I1 Details of Catalytic Polymerizations
Productivityb Temperature Polymer Catalyst Phosphine number number scavenger* Solvent Initial Max. (g PE/g Ni)
'In all caws, additives were in a 1OOO-fold excess relative to the catalyst added. bPhosphine scavenger Rh(acac)(C2H4),. In all other cases, phosphine scavenger was Ni(COD),.
TABLE IV Summary of X-ray Diffraction Data
Complex Formula NiP202C4, H4,1.5C6 H, . Fw 782.523 Space group Pi, No. 2 a (A) 12.731(5) b (4 13.680(5) c (4 12.217(5) a (Deg.) 91.94(3)
Y (Deg.1 69.34(3) v (2) 1972 p(cdcu1ated; g m-') 1.317 Cryst. dim. (MM) 0.31 X 0.30 X 0.36
Radiation Mo K, (0.71069 A) P (m-l) 6.107
Total no. unique oh. 7713
Final variables 456 R 0.037 R w 0.037
Ni(Ph PCH=C(Ph)O), 1.5C6 H, .
B (Deg.) 97.76(3)
Temp. ("C) - 100
20 limits (Deg.) 4-52
Data, > 2a(Fi) 5891
ETHYLENE HOMOPOLYMERIZATION 1995
residue was extracted with cold hexane to remove 0.1 g of a hydrocarbon wax. The remaining residue consisted of 0.45 g of Ni[Ph,PCPh=C(Ph)O], that was identified by its infrared spectrum.
X-ray Structure Determination
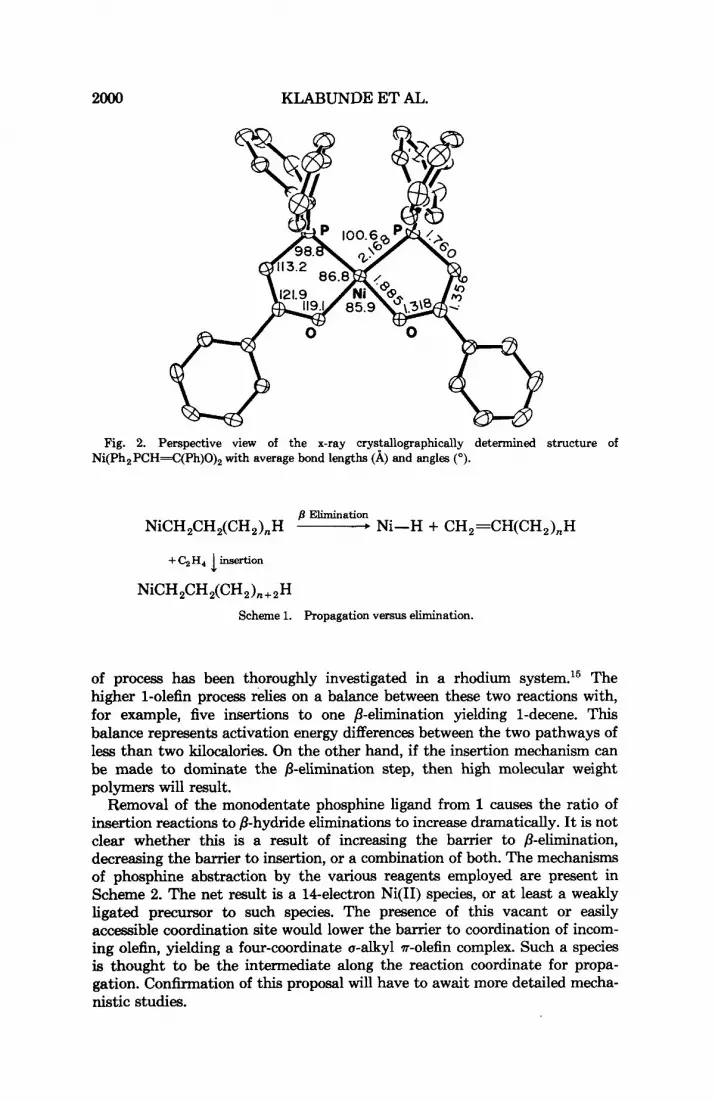
The structure of the spent catalyst, Ni(Ph,PCH=C(Ph)O),, was de- termined by standard, heavy atom techniques. Details of the experiment are presented in Table IV. Positional and thermal parameters are presented in Table V. Important bond distances and angles are presented in Figure 1 which is a perspective view of the molecule. Additional details of the structure and structure factor amplitudes can be obtained by writing to the indicated author.
RESULTS The first reaction in Table I1 clearly demonstrates the utility of the
Shell-type catalysts like 1 for the production of higher 1-olefins from ethylene in toluene. Only olefinically terminated oligomers were obtained; there was no evidence of polyethylene. Addition of (2,4-pentanedionate)-bis(ethyl- ene)rhodium(I) to the reaction mixture removes the phosphine ligand from the nickel catalyst by scavenging any dissociated ligand from solution. The effect of this is to transform a catalyst for the production of 1-olefins to a catalyst for the production of polyethylene. No oligomers are obtained; the only product is low molecular weight polyethylene. This same transformation is accomplished by adding Ni(l,BCOD), to remove phosphine.
1
Experiments 2,5,6, and 7 indicate the variety of phosphine scavengers that are effective for this reaction. Although Rh(acac)(C,H,), was found to be the best phosphine scavenger, Ni(COD), was the most attractive if catalyst lifetime is considered. Nickel carbonyl is extremely toxic and difficult to handle, so it is not a scavenger of choice. Trimethylamine oxide is a well- h0wn13 reagent for oxidative removal of ligands from metal complexes and was shown to be effective in these reactions.
Experiments 2-6, 20, and 27 indicate the wide variety of solvents in which this polymerization reaction can be run. Although hydrocarbon solvents are best, the catalysts are able to function in a variety of oxygenated solvents such as alcohols and ketones. In an attempt to better understand the effects of oxygenated solvents and other potential donor ligands on the rate of poly- merization, a series of experiments with added oxygen and nitrogen donors was carried out. Concentrations of the additives were up to lo00 times the concentrations of the catalysts but were not so great as to change the nature
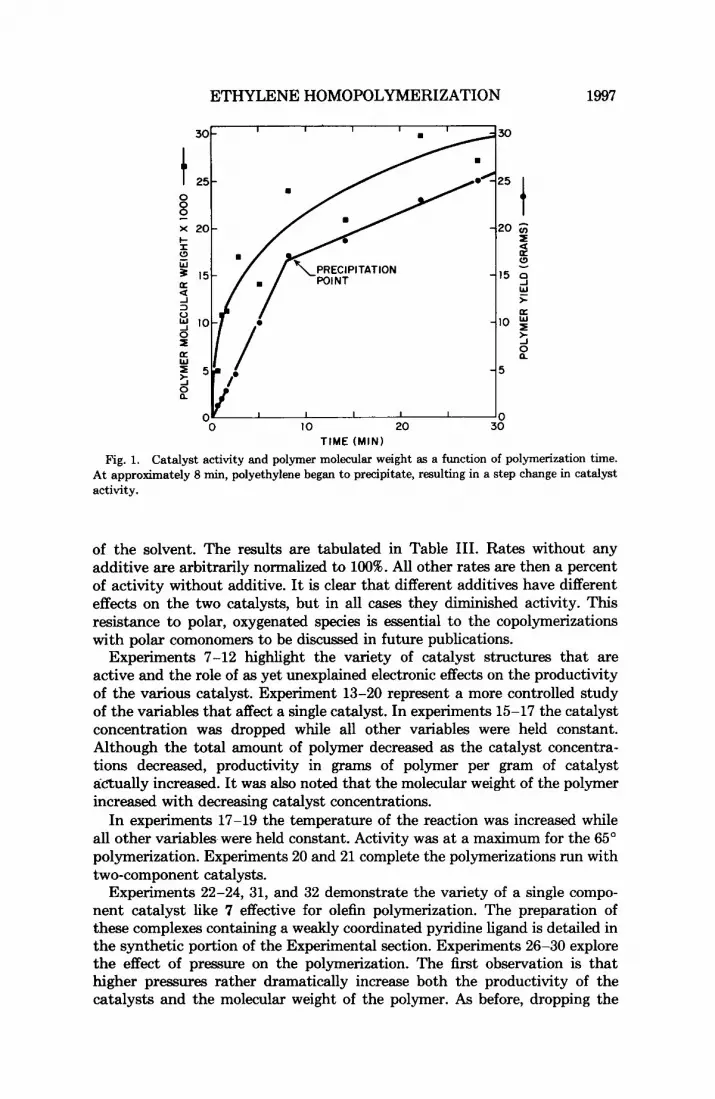
Fig. 1. Catalyst activity and polymer molecular weight as a function of polymerization time. At approximately 8 min, polyethylene began to precipitate, resulting in a step change in catalyst activity.
of the solvent. The results are tabulated in Table 111. Rates without any additive are arbitrarily normalized to 100%. All other rates are then a percent of activity without additive. It is clear that different additives have different effects on the two catalysts, but in all cases they diminished activity. This resistance to polar, oxygenated species is essential to the copolymerizations with polar comonomers to be discussed in future publications.
Experiments 7-12 highlight the variety of catalyst structures that are active and the role of as yet unexplained electronic effects on the productivity of the various catalyst. Experiment 13-20 represent a more controlled study of the variables that affect a single catalyst. In experiments 15-17 the catalyst concentration was dropped while all other variables were held constant. Although the total amount of polymer decreased as the catalyst concentra- tions decreased, productivity in grams of polymer per gram of catalyst actually increased. I t was also noted that the molecular weight of the polymer increased with decreasing catalyst concentrations.
In experiments 17-19 the temperature of the reaction was increased while all other variables were held constant. Activity was at a maximum for the 65" polymerization. Experiments 20 and 21 complete the polymerizations run with two-component catalysts.
Experiments 22-24, 31, and 32 demonstrate the variety of a single compo- nent catalyst like 7 effective for olefin polymerization. The preparation of these complexes containing a weakly coordinated pyridine ligand is detailed in the synthetic portion of the Experimental section. Experiments 26-30 explore the effect of pressure on the polymerization. The first observation is that higher pressures rather dramatically increase both the productivity of the catalysts and the molecular weight of the polymer. As before, dropping the
1998 KLABUNDE ET AL.
catalyst concentration increases productivity. Precipitation of the catalyst on Celite" diatomaceous earth increased the molecular weight of polymer. In- creasing the temperature of the polymerization to 150°C gave a solution phase process. The unsupported catalysts displayed no activity at this temperature, but a Celite"-supported catalyst remained active. The molecular weight dropped dramatically as did the molecular weight distribution of this vinyl- ended polymer.
7
Experiment 32 in Table I1 is based upon an "unsolvated)~ dimeric form of the catalyst. The unused electron pair from the oxygen of one molecule acts as a ligand in the vacant coordination site of a second molecule, resulting in a dimeric structure, 10. This structure is easily cleaved, giving the active form of the catalyst. As indicated by the polymer yield, this is a very active form of the catalyst.
H Ph
Ph H
10
Polymer molecular weight and catalyst productivity were studied as a function of time by quenching a series of polymerizations at low conversions. The results of that investigation are shown in Figure 2. Yield as a function of time is a relatively straight line until polymer begins to precipitate, carrying the catalyst out of solution. The polymerization then continues at about half the previous rate. The molecular weight of the polymer increased consistently over the initial time period, and the rate of increase diminishes at the same time that the polymerization rate decreases. A comparison of the known concentration of nickel with an estimated active center concentration indi- cated by the numbers of chains, expressed as yield divided by the number average molecular weight, revealed that even after a very short time, several chains per nickel were present. This indicates a rapid chain transfer via 8-hydride elimination.
ETHYLENE HOMOPOLYMERIZATION 1999
At the conclusion of one polymerization in which a large excess of catalyst had been employed, it was possible to isolate the spent form of the catalyst.
Ph, Ph,
This complex contains two equivalents of the P, 0-chelate ligand but no alkyl or aryl group into which olefin could insert to initiate a propagating polymer chain. It was felt that the nature of the deactivated catalyst might shed some light on the mechanism of catalyst deactivation, so an x-ray structure de- termination was carried out. The results of that determination are presented in Table V and Figure 1. Since our determination, a closely related structure has been reported.'4 Although this compound is not an active catalyst, it can be converted to an active catalyst by alkylation with an aluminum alkyl. The resulting coordinatively saturated complex, 11, contains a methyl group and two of the P, 0-chelate ligands whose oxygen atoms are bridged by a dimeth- ylaluminum group. Activity is diminished relative to its precursor and lower 1-olefins are observed, indicating that both phosphine groups are coordinated a t least part of the time. Thus, although the addition of AlR, compounds to some of the earlier polymerizations should reactivate the catalyst, it would be expected that molecular weight would suffer.
11
DISCUSSION
Phosphorus-oxygen-chelated nickel complexes of the type related to 1 are well es tab l i~hed~-~ in their role as catalyst for the conversion of ethylene to higher 1-olefins. The length of the 1-olefin chain is based upon a delicate balance between the chain propagating step, insertion of ethylene into the Ni-C bond, and the chain terminating step, 8-hydride elimination of the alkyl chain to give the 1-olefin. If the rate of 8-hydride elimination is much faster than olefin insertion, this competition, shown in Scheme 1, will result in scrambling of the olefinic hydrogen atoms with the Ni-H bond, but no useful chemistry will result. Once ethylene inserts into the Ni-H bond, it 6- eliminates back to ethylene before a second insertion can take place. This type
2000 KLABUNDE ET AL.
Fig. 2. Perspective view of the x-ray crystallographically determined structure of Ni(Ph,PCH=C(Ph)O), with average bond lengths (A) and angles (").
NiCH ,CH ,(CH , ), + ,H Scheme 1. Propagation versus elimination.
of process has been thoroughly investigated in a rhodium system.15 The higher 1-olefin process relies on a balance between these two reactions with, for example, five insertions to one &elimination yielding 1-decene. This balance represents activation energy differences between the two pathways of less than two kilocalories. On the other hand, if the insertion mechanism can be made to dominate the &elimination step, then high molecular weight polymers will result.
Removal of the monodentate phosphine ligand from 1 causes the ratio of insertion reactions to @-hydride eliminations to increase dramatically. I t is not clear whether this is a result of increasing the barrier to @-elimination, decreasing the barrier to insertion, or a combination of both. The mechanisms of phosphine abstraction by the various reagents employed are present in Scheme 2. The net result is a 14-electron Ni(I1) species, or a t least a weakly ligated precursor to such species. The presence of this vacant or easily accessible coordination site would lower the barrier to coordination of incom- ing olefin, yielding a four-coordinate o-alkyl a-olefin complex. Such a species is thought to be the intermediate along the reaction coordinate for propa- gation. Confirmation of this proposal will have to await more detailed mecha- nistic studies.
Triethylphosphine coordinates more strongly to the nickel( 11) complexes than does triphenylphosphine. Although complexes with triphenylphosphine will oligomerize ethylene to 1-olefins, we observed no reaction with their triethylphosphine analogs. Thus it appears that the stronger coordination precludes ethylene insertion. Because there are no alkyl chains, no statement can be made about the effect on 8-elimination.
Once it was clear that complete phosphine abstraction was essential to obtain high molecular weight polyethylene, our objective became to syn- thesize phosphine-free catalyst. One convenient solution was the development of weakly ligated catalysts. Coordination of pyridine ligands to the Ni(I1) catalysts is not as strong as the binding of phosphine ligands. Thus, under polymerization conditions, the pyridine ligand is largely dissociated, resulting in more active catalysts and higher molecular weight polymers. The ultimate example of this approach is the nickel dimer in which the oxygen atoms of an adjacent catalyst molecule are the readily dissociable ligand.
Ph \ /Ni\
' 2
Ph
Ph Ph
For this class of catalysts, it would be impossible to achieve a version with a more readily dissociated ligand, for if one were able to do so, it would be displaced as the h e r is reformed. As we have noted, the deactivated forms of the catalysts contain two of the
P,O-chelate ligands and no metal alkyl bond to allow polymerization. The structure, which is essentially the same as that reported elsewhere,', indicates delocalization of the bonding, with resonance between the two extremes
2002 KLABUNDE ET AL.
indicated. Treatment of these complexes with AlMe, generates catalytically active species like 11.
The role of the metal alkyl is thought to be twofold. First, it regenerates a Ni-C bond to allow propagation, but the coordination sphere of the complex is saturated, and activity would not be expected. The second role of the alkyl is thought to be removal of the second chelated ligand through the process indicated in Scheme 3. Such a process would result in the desired catalyst. The lower molecular weights from this complex may reflect incomplete coordi- nation of the phosphorus donor to the aluminum alkyl, thus allowing it to spend some time back on the nickel center as an exotic monodentate phos- phine ligand.
Me Ph,
+ Me2Alip> '0
' Al'
Scheme 3
CONCLUSION
Removal of phosphine donor ligands from nickel P, 0-chelated ethylene oligomerization catalysts yields high activity ethylene polymerization cata- lysts. These catalysts are relatively inert to materials that are effective catalyst poisons for traditional early metal-based catalysts. This has the obvious advantage of allowing processes to operate with lower purity, less expensive ethylene. The tolerance of these catalysts to oxygen donors and polar molecules is essential to their use in copolymerizations of ethylene with olefinic species containing polar functionality.
References 1. J. Boor, Ziegler-Natta Catalysts and Polymerizations, Academic Press, New York, 1979. 2. R. P. Quirk, Ed., Transition Metal Catalyzed Polymerizations-Alkenes and Dienes,
M.M.I. Press, Harwood Academic Pub., New York, 1983. 3. E. R. Freitas and C. R. Gum, Chem. Eng. Prog., Jan., 73, (1979); W. Keim, Chem. Zng.
Tech., 66,850 (1984). 4. R. Bauer, H. Chung, K. W. Barnett, P. W. Glockner, and W. Keim, US. Pat. 3,686,159
(1972); R. Bauer, P. W. Glockner, W. Keim, and R. F. Mason, US. Pat. 3,647,915 (1972); R. S. Bauer, H. Chung, P. W. Glockner, W. Keim, and H. van Zwet, US. Pat. 3,644,563 (1972); R. S. Bauer, H. Chung, P. W. Glockner, W. Keim, and H. van Zwet, US. Pat. 3,635,937 (1972).
ETHYLENE HOMOPOLYMERIZATION 2003
5. D. L. Beach and J. J. Harrison, U.S. Pat. 4,310,716 (1982); D. L. Beach and J. J. Harrison, U.S. Pat. 4,382,153 (1983); D. L. Beach and J. J. Harrison, U.S. Pat. 4,293,727 (1981); D. L. Beach and J. J. Harrison, U.S. Pat. 4,301,318 (1981); D. L. Beach and J. J. Harrison, U.S. Pat. 4,293,502 (1981).
6. K-H. A. 0. Starewsld and J. Witte, U.S. Pat. 4,537,982, Ger. Pat. Appl. to Bayer AG (1985). 7. W. Keim, F. K. Kowaldt, R. Goddard, and C. Kriiger, Angew. Chem. Znt. Ed., 17, 466
8. Y. Qingchuan, T. You& L. Huali, X. Minzhi, L. Sen, and X. Weihua, Actu Chem. Sinica,
9. W. Keim, Ann. N.Y. Acad. Sci., 415, 191 (1983).
(1978).
42, 1128 (1984).
10. D. F. Shriver, The Manipulation of Air-Sensitiue Compounds, McGraw-Hill, New York,
11. R. A. Schunn, Znorg. Synth., 13, 124 (1972). 12. R. Cramer, Znorg. Synth., 15, 116 (1974). 13. T.-Y. Luh, Coord. C h m . Reo., 60, 255 (1984). 14. H. Qichen, X. Minzhi, Q. Yanlong, X. Weihua, S. Meicheng, and T. Youqi, J. Organomet.
15. D. C. Roe, J. Am. Chem. SOC., 105, 7770 (1983).